
INTRODUÇÃO
A amiloidose é conjunto de doenças raras (afeta oito em cada 1 milhão de pessoas), progressivas e potencialmente fatais, que ocorrem quando há depósito e acúmulo em órgãos de pedaços de proteínas dobrados em uma configuração altamente estável e rígida (em folhas de pregueamento “beta”).
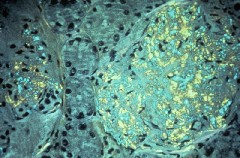
Biópsia renal corada com vermelho Congo, mostrando o aspecto “fluorescente” da proteína amilóide.
Hoje conhecemos mais de 30 proteínas que podem ser produzidas pelo organismo e depositadas dessa forma. Podem ser proteínas anormais (como na amiloidose familiar e na amiloidose sistêmica adquirida de imunoglobulinas de cadeia leve – AL), quando há uma proteína normal em excesso por longo tempo (na amiloidose sistêmica reativa – AA – e na amiloidose por beta-microglobulina associada a diálise) ou, por motivos desconhecidos, pela idade (amiloidose sistêmica senil e amiloidose do petídeo natriurético atrial).
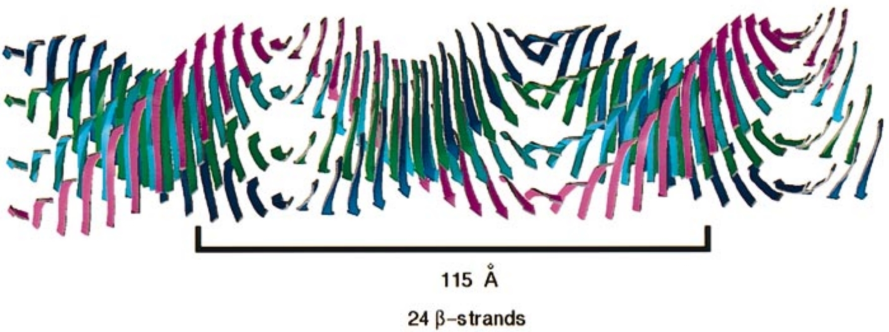
Modelo estrutural da proteína amilóide. Aqui estão representadas 4 folhas paralelas.
Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CCF J Mol Biol 1997 Oct 31;273(3):729-739
Como essas proteínas (que variam de acordo com a causa) são muito estáveis, vão se depositando em um ou mais órgãos, podendo comprometer o seu funcionamento e, em casos mais graves, morte. Os órgãos mais afetados são o coração, rins, trato gastrointestinal e sistema nervoso central, mas pode afetar também língua, músculos, pele, ligamentos, articulações, baço, pâncreas e, claro, o fígado. Apesar dos avanços no diagnóstico e tratamento nas últimas décadas, continua sendo diagnosticada geralmente apenas quando está avançada, com um terço dos portadores de AL (amiloidose de cadeia leve, a mais comum) falecendo poucos meses após o diagnóstico.
EPIDEMIOLOGIA
A amiloidose ocorre com a mesma frequência em homens e mulheres, geralmente após os 40 anos de idade. Uma revisão dos dados do National Amyloidosis Centre (NAC) de 1987 de 2012, onde são encaminhados a maioria dos casos diagnosticados no Reino Unido, mostra que a amiloidose do tipo AA (reativa sistêmica, ou secundária) diminuiu consideravelmente ao longo do tempo, provavelmente refletindo a melhora no tratamento de artropatias crônicas com o uso de medicamentos biológicos. Em compensação, aumentaram os casos de amiloidose senil, em parte pelo envelhecimento da população, mas principalmente porque o surgimento da ressonância nuclear magnética cardíaca ajudou a fazer esse diagnóstico (de fato, dados de necrópsias mostraram que um quarto dos indivíduos de mais de 80 anos possuem essa amiloidose).
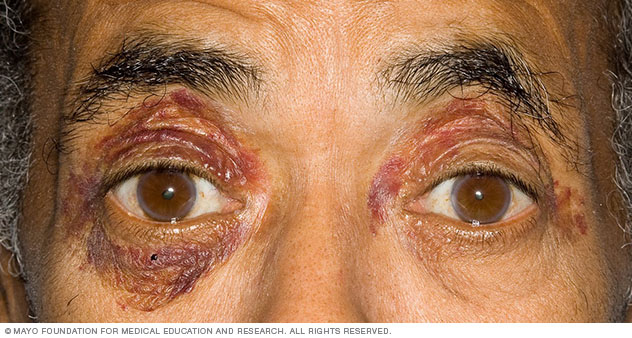
Púrpura ao redor dos olhos decorrente da fragilidade dos capilares na amiloidose (fonte)
Já a terceira amiloidose mais comum, a associada a diálise, é mais frequente no sul da Ásia, norte da África, no Oriente Médio e no México. A amiloidose hereditária é mais rara, com apenas um caso em cada 100.000 pessoas na Europa.
TIPOS DE AMILOIDOSE
Há três tipos principais de amiloidose, a primária, a secundária e a genética.
A amiloidose primária (AL) é a mais comum, onde a proteína amilóide é um fragmento da cadeia leve de imunoglobulinas (proteínas que fazem parte do sistema imunológico), que é produzida em excesso por células imunológicas. Geralmente está associada a uma doença da medula óssea, o mieloma múltiplo. O acúmulo de amilóide geralmente afeta o coração, rins, pulmões, pele, língua, nervos e intestino.
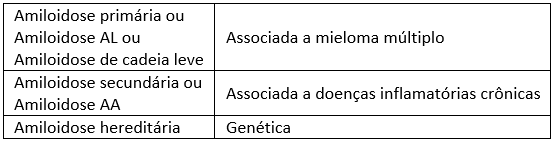
Na amiloidose secundária (AA), a proteína depositada é chamada de amilóide sérico A e é uma proteína produzida pelo fígado em resposta a processos inflamatórios. Por esse motivo, essa amiloidose é observada em pessoas portadoras de doenças inflamatórias sem tratamento adequado por mais de uma década, especialmente doenças inflamatórias intestinais (Crohn e retocolite ulcerativa), artrite reumatóide, osteomielite, tuberculose, hanseníase e outras doenças.
A amiloidose hereditária é muito rara e ocorre quando uma mutação genética leva a produção de uma proteína amilóide (transtiretina, proteína beta-amilóide P, apolipoproteína A1, procalcitonina e beta-2-microglobulina). A identificação da proteína envolvida é importante para prever suas possíveis complicações, tratamento precoce e risco de transmissão para os filhos.
SINTOMAS
Como os amiloides podem se depositar em órgãos diferentes, os sintomas e achados clínicos podem ser muito variados, como mostra a tabela abaixo.

A principal causa de mortalidade pela amiloidose é a insuficiência cardíaca, particularmente a insuficiência cardíaca direita (onde observa-se dilatação das veias jugulares no pescoço, inchaço nas pernas e no fígado), evoluindo com falência do órgão e queda na pressão arterial. Essa manifestação é rara na amiloidose secundária (reativa, AA), e mais comum na primária (de cadeia leve, AL), onde ocorre em 50% dos portadores. Além de ser mais comum na AL, o amilóide AL é tóxico às células cardíacas, fazendo com que esses pacientes tenham maior comprometimento do órgão que os das outras amiloidoses, mesmo com quantidade similar de depósito.
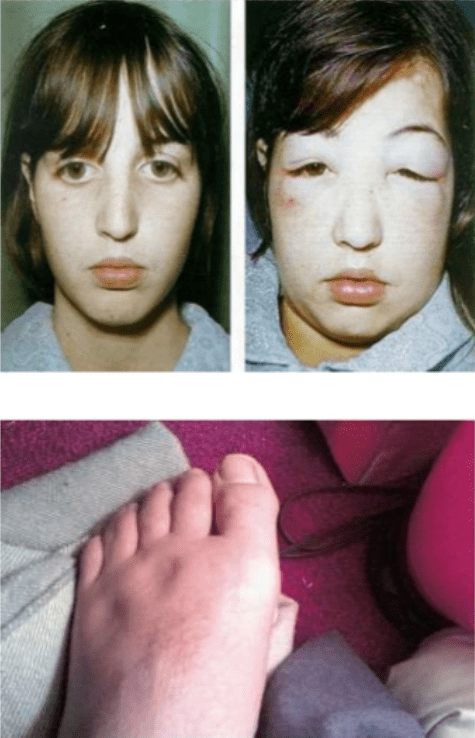
A anasarca, edema generalizado decorrente da falta de albumina (pela síndrome nefrótica, na amiloidose) se caracteriza por inchaço tanto das pernas quanto do rosto. É diferente do edema causado por problemas vasculares ou insuficiência cardíaca, por exemplo, que costuma aparecer nas pernas.
Os rins são o órgão mais afetado pelas amiloidoses AL, AA, AFib, ALect2 e AApoA1. Especialmente na AA, o depósito de amilóide provoca a chamada síndrome nefrótica, danificando o glomérulo e levando à passagem e perda de proteínas na urina. A principal proteína perdida na urina é a albumina. A albumina tem múltiplas funções, mas nesse caso específico o que mais importa é a chamada função coloido-osmótica. A albumina, como uma proteína grande, “segura” a água dentro dos vasos sanguíneos. Quando falta albumina no sangue, a água começa a vazar para os tecidos, fazendo com que as pernas inchem e comece a acumular água na barriga (ascite). Além da albumina, pode haver perda da proteína amiloide na urina, que pode ser detectada.

Tomografia computadorizada mostrando hepatomegalia em paciente com amiloidose AL (fonte)
O fígado costuma estar aumentado (hepatomegalia) pela deposição dos amiloides, mas isso raramente chega a prejudicar a sua função. Algumas vezes surge ascite, mas ela geralmente não é decorrente unicamente de hipertensão portal, mas também de insuficiência cardíaca e síndrome nefrótica.

Biópsia hepática mostrando amilóide na coloração vermelho Congo (setas) – fonte
Os nervos periféricos também podem ser afetados pelo acúmulo de amiloides, principalmente na AL. Em homens, o primeiro sintoma costuma ser a disfunção erétil (impotência), seguida de hipotensão postural (queda da pressão quando se levanta rapidamente), saciedade precoce e alteração no hábito intestinal, com diarreia, constipação ou alternando ambos. O comprometimento dos nervos sensitivos pode levar a redução na sensibilidade de extremidades ou dor.
O acúmulo de amiloide em tecidos moles pede lavar ao aumento da língua (macroglossia) e síndrome do túnel do carpo, particularmente na AL, e o histórico de doença cardíaca atípica precedida de síndrome do túnel do carpo em idosos deve levantar a suspeita de amiloidose.
DIAGNÓSTICO
Como trata-se de uma doença rara e que pode afetar múltiplos órgãos com achados clínicos variáveis, o diagnóstico pode ocorrer investigando a causa de algum comprometimento a órgão específico, mas o ideal é que seja feito pelo achado pelo conjunto de pequenos sinais e sintomas, antes que se chegue a esse ponto. E isso não é possível sem uma boa história clínica e exame físico, especialmente pelo médico generalista.
Após a suspeita clínica, pode ser realizada a pesquisa de amiloides de cadeia leve no sangue ou urina, que serve como exame de rastreamento. Depois disso, são realizadas biópsias de gordura subcutânea, mucosa retal ou labial e/ou medula óssea para confirmação diagnóstica. Em alguns casos, é necessária a coleta de biópsia do órgão afetado. Essas biópsias mostrarão o acúmulo da proteína amilóide e, se possível, identificar o tipo de amiloide, o que vai influenciar o tratamento.

Proposta de sequência diagnóstica da amiloidose por Wechalekar e colaboradores.
No fígado, além da hepatomegalia observável ao exame físico e de imagem, a biópsia revela birrefringência esverdeada no espaço de Disse após coloração com o vermelho Congo e atrofia hepatocelular (por redução do transporte de nutrientes e oxigênio), geralmente sem fibrose significativa. Como geralmente há alteração de coagulação (anormalidades vasculares, plaquetopenia, redução de fatores IX e X que se ligam às proteínas amiloides), não recomenda-se a realização de biópsia do fígado.
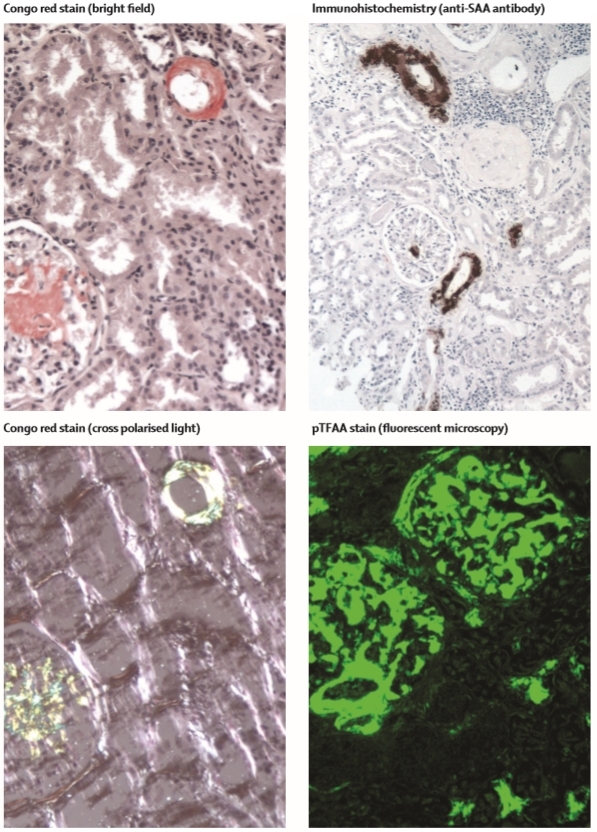
Biópsia mostrando achados típicos de amiloidose AA. A foto do canto superior esquerdo mostra o amiloide mais avermelhado na coloração de Vermelho Congo na luz normal, e o aspecto brilhante na luz polarizada (abaixo). No canto superior direito, a imunohistoquímica confirma a amiloidose AA. No canto inferior direito, a nova técnica de coloração pTFAA mostra amiloidose AL na fluorescência.
A imunohistoquímica pode demonstrar qual é o amiloide, especialmente na AA e na ATTR, mas dificilmente na AL. Outros métodos mais sofisticados incluem a pTFAA e a espectofotometria e massa do amiloide, capturado por microscópio de captura a laser.
Além da biópsia, a pesquisa do amiloide no sangue e na urina, através de eletroforese com imunofixação (IFE) consegue diagnosticar a cadeia leve da AL em 95% dos casos. Na AA, é necessário realizar biópsia de medula óssea para confirmar ou descartar mieloma múltiplo.
Em pacientes com gamopatia monoclonal de significado indeterminado (MGUS), amiloidose cardíaca e renal, frequentemente associadas à AL, a a realização da pesquisa do biomarcador da porção N-terminal do pró-hormônio do peptídeo natriurético do tipo B (NT-proBNP) pode servir como rastreamento e facilitar o diagnóstico precoce.
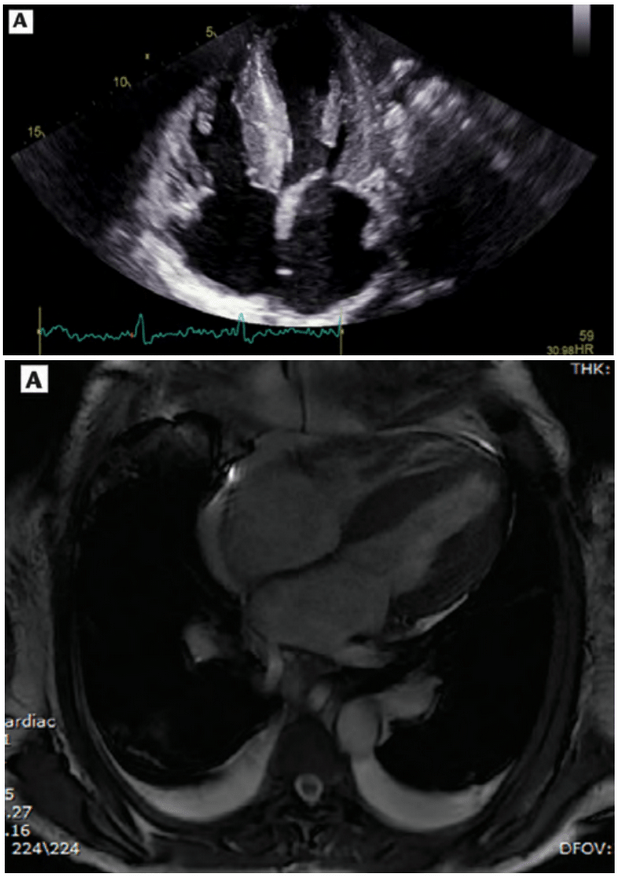
Acometimento cardíaco pela amiloidose, mostrando espessamento da parede cardíaca e redução nas cavidades no esocardiograma (acima) e ressonância (abaixo) – fonte
O próximo passo é investigar que outros órgãos estão acometidos pela doença. A cintilografia com SAP ajuda na localização de áreas afetadas, mas o grau de acumulo do amiloide por esse método tem pouca correlação com o grau de comprometimento dos órgãos, que devem então ser avaliados com os métodos mais adequados.
TRATAMENTO
Os objetivos do tratamento, a partir do diagnóstico, são reduzir ou diminuir a produção do amilóide, eliminar depósitos, aliviar ou curar a doença de base e tratar as complicações decorrentes pelos depósitos nos órgãos acometidos.
No caso das amiloidoses hereditárias ATTR, AApoAI e AFib, a fonte do amiloide é o fígado, e o transplante hepático é capaz de interromper a progressão da doença.
Na amiloidose AL, o tratamento indicado é a quimioterapia com a células monoclonal que produz o amiloide de cadeia leve como alvo. Essas células geralmente correspondem a gamopatia monoclonal de significado indeterminado (MGUS), com 10 a 15% associada a mieloma múltiplo. Os avanços significativos no tratamento do mieloma, adaptados à AL, trouxeram a uma melhora significativa no prognóstico desses pacientes, especialmente quando o transplante autólogo de células tronco é possível. Não vou me deter nos esquemas quimioterápicos existentes pois estão sempre em mudança, com novos agentes e combinações, cada um para determinada situação, e o Hematologista ou Oncologista são habilitados para propor a melhor opção para cada caso.
Na amiloidose AA, o tratamento se baseia em encontrar a doença inflamatória responsável e tratá-la. Isso pode ser feito com antibióticos na tuberculose ou agentes biológicos nas doenças reumatológicas.
CONCLUSÃO
Apesar de relativamente rara, a amiloidose é mais comum do que se geralmente reconhece se levarmos em consideração os grupos de risco, especialmente os portadores de mieloma múltiplo, gamopatia monoclonal de significado indeterminado (GMUS), idosos e portadores de doenças infecciosas e inflamatórias crônicas. Nesses, e em pacientes com sintomas ou combinações de sintomas específicos, a investigação e o tratamento adequado pode evitar complicações e melhorar tempo e qualidade de vida dos pacientes.
BIBLIOGRAFIA
- Habib, A e Neuschwander-Tetri, BA em Friedman, SJ, McQuaid, KR e Grendell, JH. Current Diagnosis & Treatment in Gastroenterology, Second Edition, McGraw Hill, 2003
- Scheuer PJ, Leftkowitch, JH; Liver Biopsy Interpretation. Saunders, 2000
- Cannon JD, Pullen RL e Rushing JD Managind the Patient with Amyloidosis, Dermatol Nurs 16(3): 255-230, 234-235, 2004
- Wechalekar AD, Gillmore JD, Hawkins PN. Systemic Amyloidosis. Lancet. 2016 Jun 25;387(10038):2641-2654. doi: 10.1016/S0140-6736(15)01274-X. Epub 2015 Dec 21. Review.
- Badar T, D’Souza A and Hari P. Recent advances in understanding and treating immunoglobulin light chain amyloidosis [version 1; referees: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):1348 (https://doi.org/10.12688/f1000research.15353.1)
- Premkumar M, Rangegowda D, Vyas T, Kulkarni A, Grover S, Mahiwall R, Sarin SK. Primary Hepatic Amyloidosis Presenting as Acute-on-Chronic Liver Failure. ACG Case Rep J. 2017 Feb 15;4:e22. doi: 10.14309/crj.2017.22. eCollection 2017. PubMed PMID: 28286788; PubMed Central PMCID: PMC5340654
- Sonthalia N, Jain S, Pawar S, Zanwar V, Surude R, Rathi PM. Primary hepatic amyloidosis: A case report and review of literature. World J Hepatol. 2016;8(6):340-4
Artigo criado em: 03/04/05
Última revisão: 03/11/18


