
INTRODUÇÃO
Descrita pela primeira vez por Jacques Caroli em 1958, é uma doença genética rara. A forma mais simples, chamada de doença de Caroli, é caracterizada como dilatação dos dutos biliares principais (maiores), sem obstrução, que podem permanecer assintomáticas até a idade adulta, e ocorre esporadicamente (sem uma forma clara de herança).
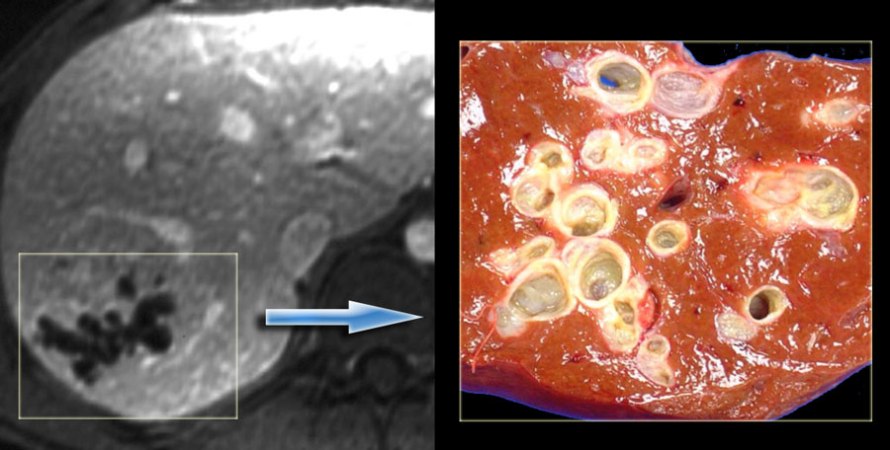
Tomografia (esquerda) e peça cirúrgica (direita) mostrando doença de Caroli bem delimitada.
Já a síndrome de Caroli se refere a uma forma mais agressiva, de herança autossômica recessiva, com dilatação de dutos biliares primários e secundário e associada a outras doenças do fígado, como a fibrose hepática congênita ou a doença policística renal autossômica recessiva (ARPKD). Quando há a presença da fibrose hepática congênita, a criança costuma manifestar sintomas decorrentes desta (hipertensão portal com esplenomegalia, ascite, edema e hemorragia por varizes esofágicas), tornando a síndrome de Caroli uma preocupação apenas secundária.
FISIOPATOGENIA
Com a dilatação das vias biliares intra-hepáticas, há acúmulo de bile e o seu fluxo fica prejudicado. Isso pode levar a formação de cálculos (pedras) de bilirrubinato de cálcio no interior dos cistos, que podem permanecer lá ou se deslocar até um ponto aonde obstruem o fluxo de bile e levam a infecção dessa (colangite) ou a obstrução do duto pancreático e provocar pancreatite.
Dilatação das vias biliares (em verde)
No entanto, mesmo sem a formação ou deslocamento desses cálculos o fluxo de bile está prejudicado e o principal sintoma é o de colangites de repetição. Colangites são infecções da bile e tendem a ser sérias, com febre, dor local, inchaço do fígado, leucocitose (aumento dos leucócitos do sangue) e icterícia (amarelão). Podem evoluir para sepse (infecção generalizada) e até a óbito se não tratadas. Podem também levar à formação de abscessos hepáticos.
QUADRO CLÍNICO E DIAGNÓSTICO
O quadro clínico da Doença de Caroli é, portanto, o de colangites de repetição. Ao investigar uma criança com esse quadro, observa-se em exames de imagem (ecografia, tomografia computadorizada e, especialmente, a colangiorressonância) a presença de dilatação sacular das vias biliares (que podem ser confundidas com doença policística hepática, pois costuma estar também associada a cistos renais). Exames laboratoriais são pouco significativos e, fora das crises de colangite, observa-se apenas leve aumento de gama-glutamiltransferase e fosfatase alcalina. A colangiopancreatografia endoscópica retrógrada e a colangiografia transparietal, onde são injetados contrastes (por via endoscópica e por punção através da pele, respectivamente) estão formalmente contra indicadas pois podem levar a retenção do contraste e colangite severa.
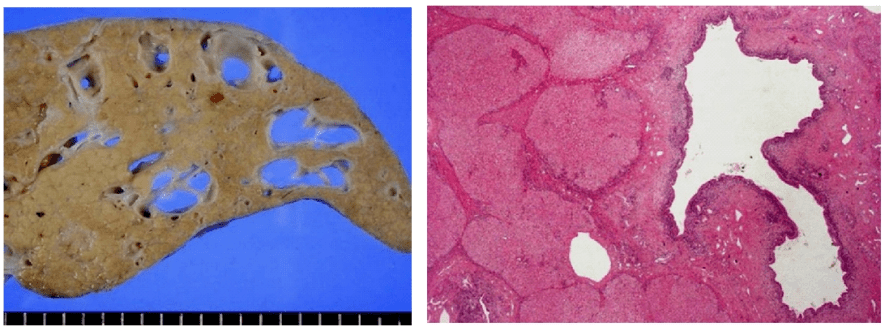
Paciente com síndrome de Caroli acompanhada de fibrose hepática congênita, com cistos visíveis no fígado a olho nu (esquerda) e na biópsia (direita) – fonte
Os diagnósticos diferenciais (outras doenças semelhantes que precisam ser descartadas) incluem colangite esclerosante primária, cistos de colédoco, doença policística hepática isolada, cistos hepáticos e hamartomas biliares.
TRATAMENTO
O tratamento básico consiste no diagnóstico e tratamento precoce das crises de colangite. Além do risco de cada infecção, com o tempo pode se desenvolver amiloidose e insuficiência hepática. Se houver sinais de obstrução por cálculo, este precisa ser retirado. Um modo de se prevenir o aparecimento desse cálculo é o uso contínuo de ácido ursodeoxicólico.

Tomografia computadorizada mostrando dilatações biliares com o “sinal do ponto central” (setas), que correspondem a veias portais e artérias hepáticas (fonte)
Outra grande preocupação é o surgimento de câncer de vias biliares (colangiocarcinoma), pois o risco de desenvolver esse tipo de câncer é 100 vezes maior em quem tem Doença de Caroli do que nas demais pessoas.
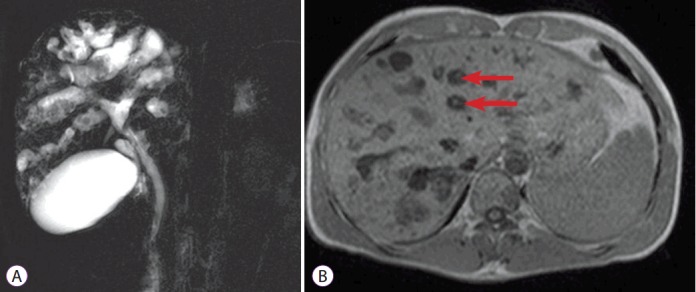
Colangiorressonância mostrando múltiplas dilatações irregulares com o típico “sinal do ponto central” e presença de cálculos (fonte)
Se as dilatações forem limitadas em uma pequena área do fígado, pode-se realizar cirurgia para retirada dessa área, com baixa morbidade (complicações) e baixíssima mortalidade. Uma outra boa opção de tratamento é o transplante de fígado se houver progressão para doença grave, com insuficiência hepática, após colangite grave (se não houver possibilidade de ressecção parcial) ou na suspeita de colangiocarcinoma.
BIBLIOGRAFIA
- Zetterman, RK em Friedman, SJ, McQuaid, KR e Grendell, JH. Current Diagnosis & Treatment in Gastroenterology, Second Edition, McGraw Hill, 2003
- Bazlul Karim, ASM. Caroli´s Disease. Indian Pediatrics, 41: 848-850, 2004 ( link )
- Sherlock, S e Heathcote, J em Bircher, J, Benhamou, JP et al. Oxford Textbook of Clinical Hepatology, Oxford Medical Publications,1999.
- Habib S ; Shakil O ; Couto OF ; Demetris AJ ; Fung JJ ; Marcos A ; Chopra K Caroli’s disease and orthotopic liver transplantation. Liver Transpl.12(3):416-21, 2006
- Bockhorn M ; Malag M ; Lang H ; Nadalin S ; Paul A ; Saner F ; Frilling A ; Broelsch CE The role of surgery in Caroli’s disease. J Am Coll Surg. 202(6):928-32, 2006
- De Kerckhove L; De Meyer M; Verbaandert C; Mourad M; Sokal E; Goffette P; Geubel A; Karam V; Adam R; Lerut J The place of liver transplantation in Caroli’s disease and syndrome. Transpl Int. 19(5):381-8, 2006
- Kassahun WT ; Kahn T ; Wittekind C ; Mssner J ; Caca K ; Hauss J ; Lamesch P Caroli’s disease: liver resection and liver transplantation. Experience in 33 patients. Surgery. 138(5):888-98, 2005
- Caroli Disease – Orphanet (link)
- Yasunori Sato, Xiang Shan Ren, and Yasuni Nakanuma, “Caroli’s Disease: Current Knowledge of Its Biliary Pathogenesis Obtained from an Orthologous Rat Model,” International Journal of Hepatology, vol. 2012, Article ID 107945, 10 pages, 2012. https://doi.org/10.1155/2012/107945
-
Hwang MJ, Kim TN. Diffuse-Type Caroli Disease with Characteristic Central Dot Sign Complicated by Multiple Intrahepatic and Common Bile Duct Stones. Clinical Endoscopy. 2017;50(4):400-403. doi:10.5946/ce.2016.150.
- Yonem O, Bayraktar Y. Clinical characteristics of Caroli’s syndrome. World Journal of Gastroenterology : WJG. 2007;13(13):1934-1937. doi:10.3748/wjg.v13.i13.1934.
Artigo criado em: 17/04/05
Última revisão: 17/10/18


