
Dra. Adriana Maria Alves De Tommaso
Dr. Stéfano Gonçalves Jorge
INTRODUÇÃO
- Descrita, pela primeira vez, em 1912, pelo Dr. Samuel Alexander Kinnier Wilson, também denominada degeneração hepatolenticular;
- É um distúrbio primário do metabolismo do cobre, causado pela mutação de gene ATP7B, responsável pelo transporte deste metal, levando ao seu acúmulo, inicialmente no hepatócito e posteriormente em diversos órgãos e tecidos, particularmente no cérebro, córnea e rins;
- Metabolismo do cobre: a dieta normal de uma pessoa contém cerca de 2 a 5 mg de cobre por dia, sendo necessário (pois o cobre é essencial para o nosso metabolismo) apenas 0,9 mg – o excedente precisa ser eliminado; o cobre, absorvido no intestino, é transportado ao fígado ligado à albumina e à histidina, sendo avidamente removido da circulação pelos hepatócitos, que o liga à apotioneína para formar a Cu-metalotioneína ou o incorpora à ceruloplasmina, retornando à circulação, ou é excretado na bile. Os dois últimos passos estão alterados na Doença de Wilson. Na bile, o cobre liga-se a diferentes substâncias, estando associado a substâncias derivadas ou semelhantes à ceruloplasmina;
- Incidência ( doença= 30:1000000, portador heterozigoto= 1:100);
- Herança autossômica recessiva => cromossomo 13, locus 13ql4.3 => ATP7B => excreção biliar de cobre está marcadamente diminuída e o metal não pode ser incorporado adequadamente à ceruloplasmina. Consequentemente, o cobre em excesso distribui-se inicialmente no citoplasma, provavelmente ligado a uma forma não-tóxica de metalotioneína, e posteriormente aparece em densos grânulos nos lisossomos. O cobre livre (não ligado à ceruloplasmina), liberado na circulação a partir de hepatócitos lesados, passa então a se acumular em diversos órgãos;
- Mais de 300 mutações descritas.
QUADRO CLÍNICO
- Variável;
- Habitualmente no final da infância e na adolescência;
- Cerca de 40% inicia-se com doença hepática, várias formas de apresentação (hepatite crônica ativa, hepatite fulminante, cirrose assintomática, elevação de transaminases);
- Insuficiência hepática fulminante => sinais de alerta => anemia hemolítica com Coombs negativo, FALC baixa em contraste com altos níveis de bilirrubina, pouca elevação de transsaminases (sendo AST>ALT);
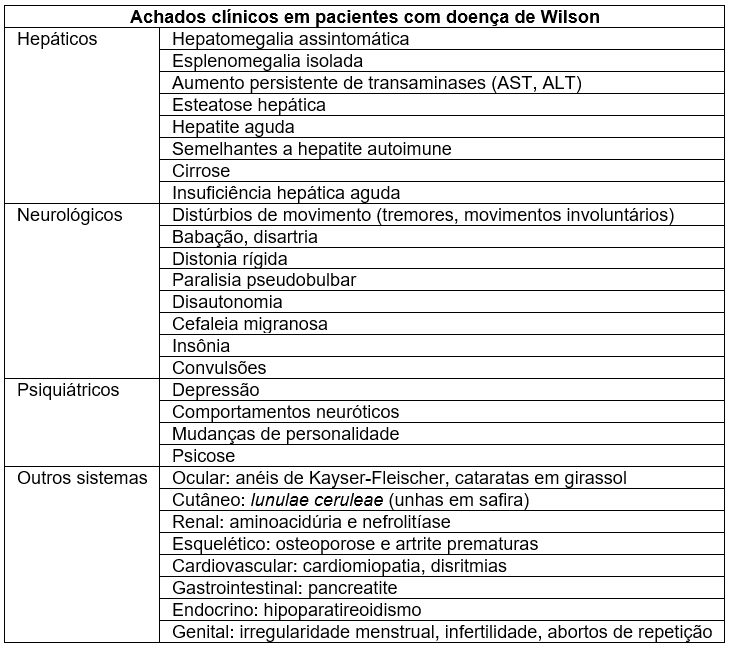
- Manifestações neurológicas: as estruturas mais atingidas são os gânglios da base. Podem também estar acometidos o córtex cerebral, a substância branca subcortical, o tálamo, o núcleo subtalâmico, a substância negra e o cerebelo. Quadro Clínico: As anormalidades neurológicas são predominantemente motoras e representadas por distúrbios do movimento tais como distonia, diversos tipos de tremor (postural ou de repouso), rigidez, bradicinesia, Coréia, atetose, ataxia e instabilidade postural. A fala e a marcha estão frequentemente afetadas. A quase totalidade dos pacientes com quadro neurológico, apresenta cirrose estabelecida. Há ampla faixa de idade de aparecimento dos sintomas (entre 8 e 50 anos). Na maioria, a idade de apresentação encontra-se na primeira e segunda décadas de vida, sendo excepcional acima de 40 anos;
- Manifestações oftalmológicas: a mais comum e importante é o anel de Kayser-Fleisher, que se forma na membrana de Descemet. Pode estar ausente em até cerca de 50% dos casos com manifestações exclusivamente hepáticas, de instalação na infância ou na adolescência. Como regra pode-se afirmar que nas formas neurológicas o anel está sempre presente;
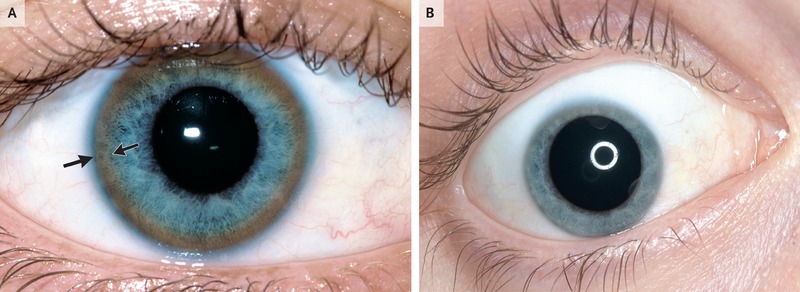
Paciente de 18 anos com doença de Wilson e anel de Kayser-Fleisher (setas, à esquerda), com resolução completa após tratamento com quelação do cobre (direita) – fonte
- Manifestações osteoarticulares: geralmente relacionadas à lesão tubular renal com perda de cálcio e fósforo, a mais comum é a osteoporose, que pode determinar fraturas espontâneas. Outros tipos de acometimento são osteomalácia, osteoartrite e osteocondrite dissecante;
- Manifestações renais: predominam as decorrentes de lesão tubular, tais como hiperaminoacidúria, hiperfosfatúria, hipercalcíuria renal e hiperuricosúria;
- Manifestações hematológicas: hiperplenismo, relacionado à hipertensão portal, e a anemia hemolítica, decorrente de altos níveis de cobre sérico livre;
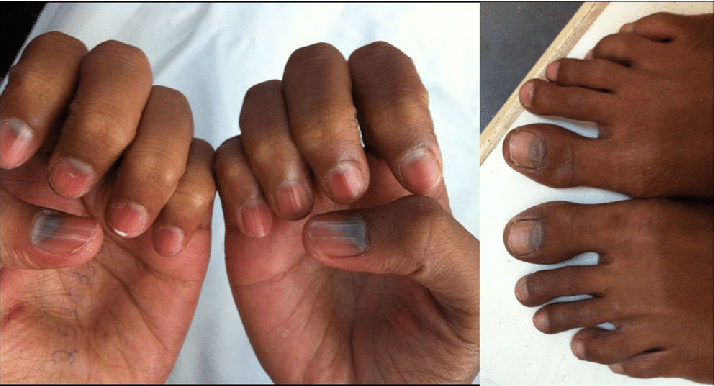
lunulae ceruleae ou lúnula azulada (também chamada de unhas em safira) – fonte
- Manifestações dermatológicas: sem gravidade, podem estar presentes sob a forma de hiperpigmentação nos membros inferiores, lúnula azulada e acantose nigricans.
DIAGNÓSTICO
- Não há um único teste => clínica, bioquímica, história familiar;
- Ceruloplasmina: níveis séricos baixos são encontrados em 90% a 95% dos pacientes. Por outro lado, 20% dos heterozigotos têm níveis séricos baixos de ceruloplasmina, mas não evoluirão com a doença. Valores baixos também são encontrados em outras situações;
- Anel de Kayser-Fleischer: é visto quando há manifestações neurológicas. Por outro lado, quando há lesão hepática, está presente em 55-70% casos;
- Cobre sérico total: avalia, também, o cobre sérico ligado à ceruloplasmina. Portanto, se esta estiver com níveis muito baixos, o cobre total estará diminuído;
- Cobre urinário: quase todos os pacientes sintomáticos apresentarão níveis elevados (>100mcg/d). Também é importante para acompanhar a aderência ao tratamento, bem como a eficácia da medicação. Pode ocorrer em outras doenças hepatobiliares. Sensibilização com a D-penicilamina é importante e auxilia a diminuir os resultados falso negativos;
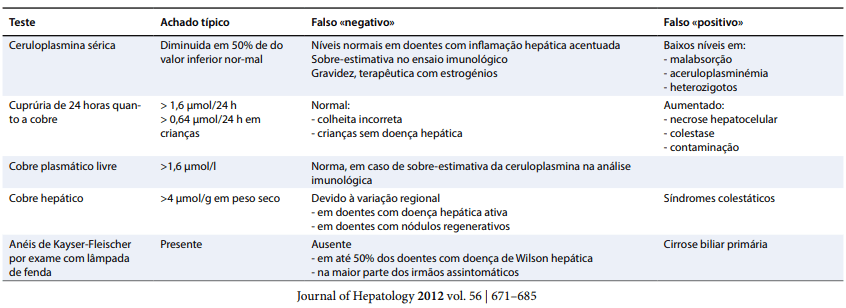
- Dosagem de cobre no tecido: padrão ouro. Concentração normal = 20-40 mcg/g de peso seco. Na doença, costuma ultrapassar 250 mcg/g, mas isso também pode ocorrer em doenças colestáticas. Outro problema é que na doença hepática avançada a distribuição de cobre é irregular no fígado, e uma biópsia pode ser realizada em uma área com menor concentração de cobre. Mas a principal limitação da biópsia é que não é todo laboratório que faz essa dosagem. Se essa investigação for realizada, a biópsia deve ser colhida por aspiração ou agulha de corte nova, colocada em frasco livre de cobre seco e congelado imediatamente ou colocado em forno para secagem à vácuo;
- Coloração rodanina: tecido hepático. Presente em outras doenças colestáticas;
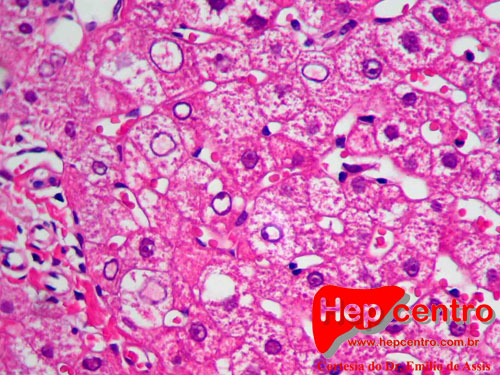
- Pesquisa de cobre com radioisótopo: há diminuição da incorporação de cobre radioativo pela ceruloplasmina. Os testes oferecem grandes dificuldades técnicas para sua execução e por essa razão são pouco utilizados. Ademais existe uma sobreposição considerável entre heterozigotos e pacientes;
- Diagnóstico genético: como há mais de 500 mutações diferentes, além de outras 100 não patogênicas, o diagnóstico genético é difícil, pouco prático e acaba sendo reservado para familiares de pacientes com Doença de Wilson com mutação específica já diagnosticada. No Brasil, em estudo realizado por Deguti e colaboradores, as mutações mais comuns envolveram os exons 8 e 15. Como vantagem sobre as demais, permite o diagnóstico logo no primeiro ano de vida;
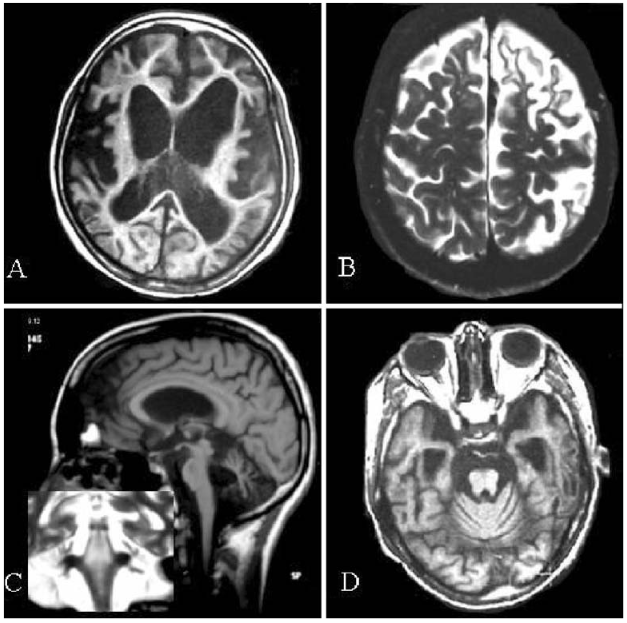
Achados típicos de doença de Wilson em ressonância nuclear magnética. A: criança de 14 anos com atrofia cortical e subcortical difusas. B: mulher de 26 anos com atrofia frontal e parietal focais à esquerda. C: moça de 15 anos com atrofia cerebelar e de tronco cerebral. D: rapaz de 16 anos com atrofia cerebelar e de tronco cerebral, com atrofia dos lobos temporais e dilatação dos ventrículos laterais (fonte)
- Ressonância magnética: parece ser sensível para detectar alterações precoces em pacientes com a doença. Mostram alterações localizadas com predileção para os gânglios da base nos casos com manifestações neurológicas. Essas anormalidades embora não sejam específicas, são altamente sugestivas. Um sinal considerado característico é o chamado “face do panda gigante” (mesencéfalo). Outra alteração recentemente descrita em pacientes com sintomas neurológicos é a presença do claustrum brilhante.
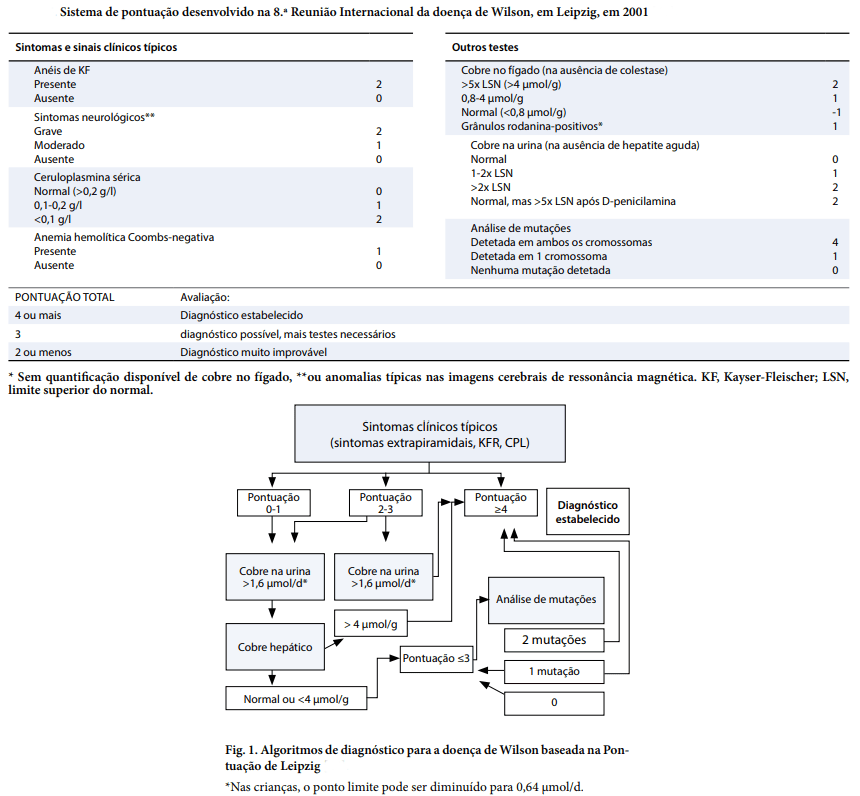
- Pela dificuldade de se fechar o diagnóstico, foi proposto em 2001 em Zeipzig uma sequencia de investigação e score
TRATAMENTO
Se não tratada, a doença é universalmente fatal, mais frequentemente pela doença hepática e menos por neuropatia progressiva. O objetivo é promover o balanço negativo de cobre, com a finalidade de remover os depósitos anormais desse metal no organismo. Com essa finalidade, as estratégias de tratamento empregadas são:
- Redução da ingestão de cobre;
- Aumentar a excreção de cobre (principal forma de tratamento).
Dieta
- Com os meios farmacológicos disponíveis atualmente, restrições dietéticas drásticas são dispensáveis, mas recomenda-se mesmo assim diminuir o consumo de alimentos ricos em cobre, como chocolate, “miudos”, nozes, legumes, frutos do mar, frango, cogumelos e frutas secas. O ideal é ingerir menos de 0,6mg/dia (a dieta normal tem de 1 a 5 mg/d).
Agentes quelantes
- D-penicilamina (distribuída no Brasil pela Merck Sharp & Dohme, com o nome de cuprimine®). Tem sido usada desde 1956 e é a forma mais importante de quelação do cobre, com aumento da excreção urinária. A dose para crianças é de 20mg/kg/d, divididas em 2 a 4 vezes ao dia. Para adultos a dose varia de 750 a 1.500 mg/d (cada comprimido tem 250 mg) divididos em 2 a 3 vezes ao dia. A medicação deve ser tomada pelo menos 1 hora antes das refeições e, como causa redução da ação de piridoxina essa deve ser suplementada (25-50mg/d). Plaquetopenia e leucopenia são complicações importantes, ocorrendo também anemia aplástica e agranulocitose. Toxidade renal (proteinúria e hematúria) é reversível. Em algumas situações, mesmo após a suspensão da droga, ocorre progressão para síndrome nefrótica e glomerulonefrite membranosa. Em caso de piora dos sintomas neurológicos, o tratamento consiste na redução da dose da medicação. Pode ser necessária a troca da medicação por outro agente quelante ou sais de zinco, todavia as manifestações neurológicas nem sempre regridem. A medicação é segura durante a gravidez.
- Trientine (trietilenotetramina): do mesmo laboratório, nome comercial= Syprine®, ainda não disponível no Brasil. É uma alternativa bem razoável para os pacientes com reações colaterais importantes à D-penicilamina. Também aumenta a excreção urinária, porém é menos potente. É efetivo por via oral, com dose máxima de 2 g por dia para adultos e 1,5 g para crianças, divididas em duas a quatro tomadas, em jejum. Infelizmente, pacientes que apresentam reações adversas à D-penicilamina, como formas graves de lúpus e lesões renais, podem também ser suscetíveis ao trientine.
- Acetato ou Sulfato de Zinco: age bloqueando a absorção de cobre pelo trato intestinal. Sua maior vantagem é a ausência de efeitos colaterais. O zinco induz a síntese de metalotioneína (proteína carreadora de metais que oferece sítios de ligação para o cobre livre). Acredita-se que a diminuição da absorção do cobre deva-se à indução da metalotioneína intestinal , que se ligaria ao cobre absorvido na mucosa intestinal. Em consequência à descamação da mucosa intestinal, o cobre seria eliminado nas fezes. Poderia também ocorrer indução de uma metalotioneína hepática, que transformaria o cobre tissular em não-tóxico. Os sais de zinco devem ser reservados para tratamento de manutenção, após o paciente ter sido eficazmente tratado com quelantes de cobre. Há quem o considere indicado no tratamento inicial de pacientes assintomáticos e de mulheres grávidas. A dose de sulfato de zinco é de cerca de 200 mg, três vezes ao dia (75-300 mg/dia do zinco elemento), 30-60 minutos antes das refeições. A administração com o estômago vazio justifica-se porque muitas substâncias presentes na dieta previnem a eficácia da medicação. O efeito colateral mais comumente observado com o uso do sal sulfato é a irritação gástrica, que pode ser contornada com sua substituição pelo sal acetato, que é melhor tolerado e deve ser empregado também em três doses diárias de 170 mg cada.
- O tetratiomolibdato ainda está em fase experimental, mas é especialmente promissor em pacientes com manifestações neurológicas. Se administrado com a refeição, forma complexos de cobre não absorvíveis. Se administrado em jejum, forma complexos com cobre presente no sangue que são depositados no fígado, mas esses complexos são inativos e não causam doença.

Terapia celular: ainda em estágio experimental, o transplante de células do próximo paciente com atividade normal da ATP7B (após correção do gene por técnica como CRISP9) pode implicar na resolução da função hepática e interromper a progressão da doença neurológica sem a necessidade da imunossupressão pós transplante hepático.
Transplante Hepático: reservado para os casos onde não há resposta ao tratamento, ou nos casos onde a apresentação inicial já é o de uma falência hepática. Como o fígado transplantado tem atividade normal da ATP7B, não há progressão da doença, mas as lesões neurológicas se mantém e há necessidade da imunossupressão pelo transplante.
BIBLIOGRAFIA
-
EASL Clinical Practice Guidelines: Wilson’s disease. European Association for the Study of the Liver, Journal of Hepatology , Volume 56 , Issue 3 , 671 – 685, 2012 (link – em português)
- Hedera P. Update on the clinical management of Wilson’s disease. The Application of Clinical Genetics. 2017;10:9-19. doi:10.2147/TACG.S79121. (link)
- BMJ Clinical Practice: Wilson Disease
- Koh C, Zhao X, Samala N, Sakiani S, Liang TJ, Talwalkar JA. The AASLD Clinical Practice Guidelines: A Critical Review of Scientific Evidence and Evolving Recommendations. Hepatology (Baltimore, Md). 2013;58(6):2142-2152. doi:10.1002/hep.26578 (link)
- Ferenci, P. , Caca, K. , Loudianos, G. , Mieli‐Vergani, G. , Tanner, S. , Sternlieb, I. , Schilsky, M. , Cox, D. and Berr, F. (2003), Diagnosis and phenotypic classification of Wilson disease. Liver International, 23: 139-142. doi:10.1034/j.1600-0676.2003.00824.x
- Sinha S, Taly AB, Ravishankar S, et al. Wilson’s disease: cranial MRI observations and clinical correlation. Neuroradiology. 2006;48(9):613–621
- Deguti MM, Genschel J, Cancado EL, et al. Wilson disease: novel mutations in the ATP7B gene and clinical correlation in Brazilian patients. Hum Mutat 2004;23:398
- Filippi, C. and Dhawan, A. (2014), Current status of human hepatocyte transplantation and its potential for Wilson’s disease. Ann. N.Y. Acad. Sci., 1315: 50-55. doi:10.1111/nyas.12386
Artigo criado em: 2003
Última atualização: 03/06/2020


