
Dra. Adriana Maria Alves De Tommaso
INTRODUÇÃO
- Doença genética rara do metabolismo da galactose.
- Defeito em uma das enzimas responsáveis pela conversão da galactose em glicose e, dependendo da enzima defeituosa, a doença se manifesta de maneira diferente.
- Gene descoberto em 1956.
- É classificada em galactosemia do tipo 1, 2, 3 e a Duarte.
- Via metabólica:

GALACTOSEMIA TIPO I
- Deficiência no metabolismo da galactose por deficiência da enzima galactose-1-fosfato uridil transferase.
- O portador dessa deficiência pode ter danos causados aos rins, ao fígado, ao cérebro e aos ovários.
- A incidência é de 1:50.000.
- Preferencialmente neonatos (assim que se introduz a lactose na sua dieta).
- Esse acúmulo de galactose-1-fosfato causa danos às células parenquimais dos rins e fígado, cérebro, ovários e olhos:
- Fígado => Cirrose
- Rins => Síndrome de Fanconi
- Cérebro => Retardo mental
- Ovários => Amenorréia 1ária ou 2ária
- Olhos => Catarata
- Os danos podem se iniciar na fase pré-natal a partir da galactose transplacentária vinda da mãe heterozigota.
- O excesso de galactose é convertido em galactitol, um álcool que é o responsável pela catarata.
- O acúmulo de galactose-1-fosfato é a causa dos danos aos ovários
- Principais manifestações:
- Hepáticas e gastrointestinais: irritabilidade, letargia, vômitos, dificuldade de alimentação, baixo ganho de peso, hepatomegalia, esplenomegalia, ascite, cirrose hepática.
- Neurológicas: retardo mental, problemas de fala e coordenação motora.
- Endócrinas: disfunção ovariana com amenorréia 1ária ou 2ária, provável aumento de risco de câncer ovariano.
- Oculares: catarata.
- Infecciosas: a galactose inibe a atividade anti-bacteriana dos leucócitos e isso aumenta a freqüência de mortes neonatais por infecção por E. coli.
GALACTOSEMIA TIPO II
- Deficiência da atividade da galactoquinase.
- A incidência é de 1:40.000 crianças.
- Danos principalmente aos olhos.
- As características clínicas do portador desse tipo de Galactosemia são a presença de catarata e inteligência normal.
GALACTOSEMIA TIPO III
- Alteração da atividade da uridil difosfo galactose-4-epimerase resultando em duas formas da doença: inicial e grave.
- Incidência muito rara.
- Pode ocorrer desde o recém nascido ou no decorrer da infância.
- Forma inicial: pacientes assintomáticos. A enzima deficiente apenas nas células do sangue, sendo normal nos outros tecidos. A descoberta casual em teste neonatal onde a galactose-1-fosfato está elevada.
- Forma grave: pacientes com sintomas clínicos idênticos à forma clássica da Galactosemia (tipo 1). A enzima deficiente nas células sangüíneas e também nos fibroblastos ( onde sua atividade é menor que 10% do normal).
GALACTOSEMIA DUARTE
- Existem 2 formas diferentes: Duarte-1 (atividade 150% superior à normal) e Duarte-2 (50% da atividade normal). Muitas crianças possuem a variante clássica (herdada de um pai) e a variante Duarte (herdada do outro pai) e são chamadas variantes DG, com atividade da enzima entre 14 e 25% do normal.
- Diagnóstico nas primeiras semanas de vida, com atividade da GALT de 25-50%.
- Controvérsia com relação à restrição dietética. Alguns serviços fazem restrição até 1 ano => reintrodução gradativa com dosagem da enzima. Em estudo recente (2017), Lynch e colaboradores, ao avaliar 10 crianças com galactosemia DG detectaram sutis, mas déficits de desenvolvimento potencialmente problemáticos. Talvez a restrição deva se extender além de 1 ano de díade, mas ainda são necessários mais estudos.
Diagnóstico:
- Pesquisa de substância redutoras na urina (exame de triagem).
- Dosagem da enzima.
- Biópsia hepática.
- O teste de tolerância à galactose é contra-indicado, uma vez que pode ser fatal ao paciente portador de Galactosemia.
Tratamento:
- Dieta livre de galactose e lactose.
- Mais intensa no tipo 1 e na forma grave do tipo 3.
- Monitoramento constante do nível de galactose e de seus metabólitos no sangue.
- Monitoramento multidisciplinar.
TRATAMENTO
- Dieta livre de galactose e lactose.
- Mais intensa no tipo 1 e na forma grave do tipo 3.
- Monitoramento constante do nível de galactose e de seus metabólitos no sangue.
- Monitoramento multidisciplinar.
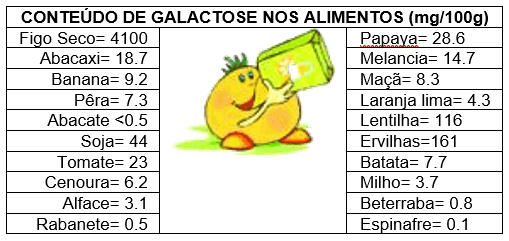
DIETA

PROGNÓSTICO
- Varia em função do tipo de Galactosemia.
- Galactosemia tipo 1 e forma grave da Galactosemia tipo 3: mesmo que o tratamento seja realizado corretamente, deve ocorrer na criança uma baixa intelectualidade, problemas de fala e disfunções ovarianas nas meninas.
- Galactosemia tipo 2: a dieta rígida pode reverter cataratas formadas por causa da doença.
- A forma inicial da Galactosemia tipo 3, por sua vez, apresenta bom prognóstico caso seja realizado seriamente o tratamento previsto por um diagnóstico precoce.
Artigo criado em 12/11/06
Última revisão: 21/06/2020


