

INTRODUÇÃO
A síndrome de Gilbert é uma condição (não uma doença!) causada por mutações genéticas. Essas prejudicam o funcionamento de uma enzima no fígado, levando ao aumento da bilirrubina em algumas condições, como jejum prolongado, medicamentos, infecções e atividade física intensa. Nessas situações, a pessoa fica levemente amarelada, especialmente nos olhos.

É uma condição, não uma doença em si, porque não compromete nenhum órgão, não leva a complicações e os sintomas tendem a surgir na adolescência e desaparecer alguns anos depois. É muito comum, sendo observada em 4 a 16% das pessoas, sendo que apenas 30% apresentam a icterícia.
O QUE OCORRE NO ORGANISMO ?
A síndrome de Gilbert é caracterizada pelo aumento da bilirrubina indireta no sangue, causada por uma enzima que funciona mal. É bem simples, mas para entender precisamos saber o mínimo sobre o metabolismo da bilirrubina. Só o mínimo, prometo ser breve.
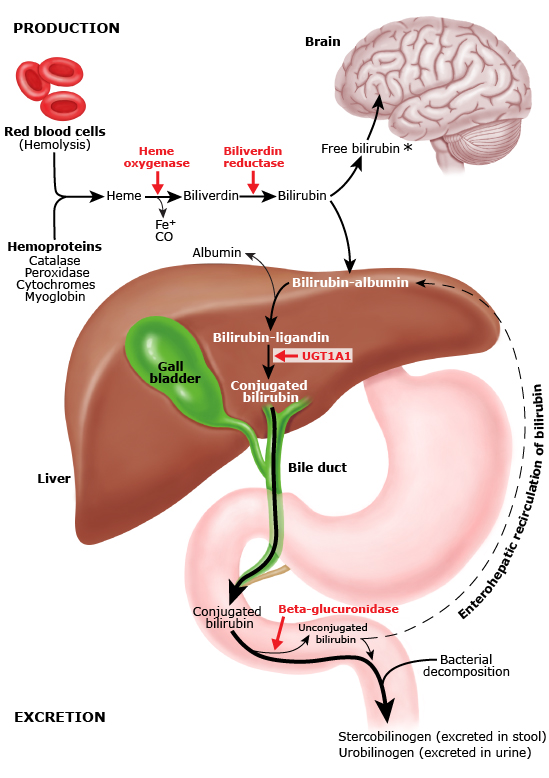
O metabolismo da bilirrubina começa com as hemácias, as células vermelhas do sangue. Essa células são abarrotadas de hemoglobina, aquela proteína que carrega oxigênio e gás carbônico dos pulmões para todo o organismo, e vice versa. Acontece que as hemácias tem seu “prazo de validade” ao redor de 120 dias. Quando ficam velhas, são destruídas no baço e as hemoglobinas no seu interior são degradadas em biliverdina e depois em bilirrubina indireta (também chamada de “não conjugada”), que chega no fígado diretamente pela veia esplênica.
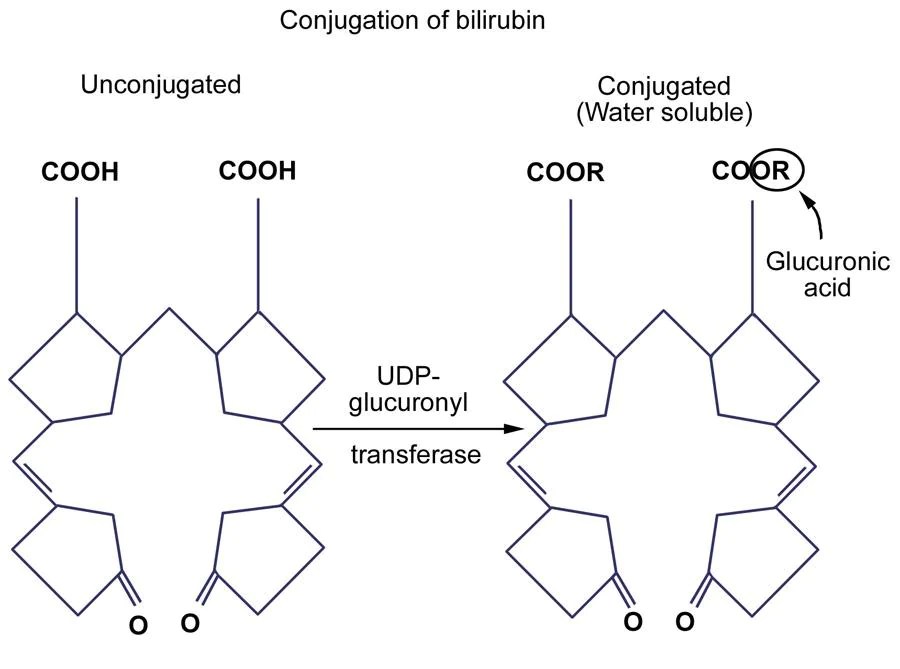
Acontece que essa enzima é codificada por um gene chamado de UGT1A1. Esse gene pode ter diversos erros diferentes (já foram observadas mais de 130 mutações!), que reduzem a sua função em até 30%. Essa porcentagem é suficiente para a pessoa viver muito bem, mas em algumas situações ou há excesso de bilirrubina indireta para conjugar, ou uma série de fatores reduz mais ainda a atividade da enzima. Então começa a haver acúmulo de bilirrubina indireta e a icterícia aparece. Quando esses fatores se resolvem, a icterícia desaparece sem deixar nenhuma consequência.

Essas mutações, para causar a síndrome, precisam ocorrer em homozigose, ou seja, você precisa herdar um gene mutante da mãe e outro do pai, o que torna essa condição como de herança autossômica recessiva (para quem gosta de genética). Parece muito improvável ? Acontece que 42% das pessoas tem um dos genes com mutação, e 9% tem os dois ! Mas só cerca de 1/3 das pessoas com ambos os genes vão ter icterícia alguma vez.
SINTOMAS
Como a síndrome causa aumento leve da bilirrubina indireta, o único sintoma claramente relacionado à síndrome é a icterícia, que por ser leve geralmente é observada apenas nos olhos. Outros sintomas que costumam ser relatados, como dor abdominal, fraqueza, náuseas e mal estar, podem não tem nenhuma relação com a síndrome. Afinal, muitas vezes a síndrome é descoberta investigando sintomas que não tem nada a ver com ela, por pessoas que mal notavam a icterícia antes do diagnóstico.
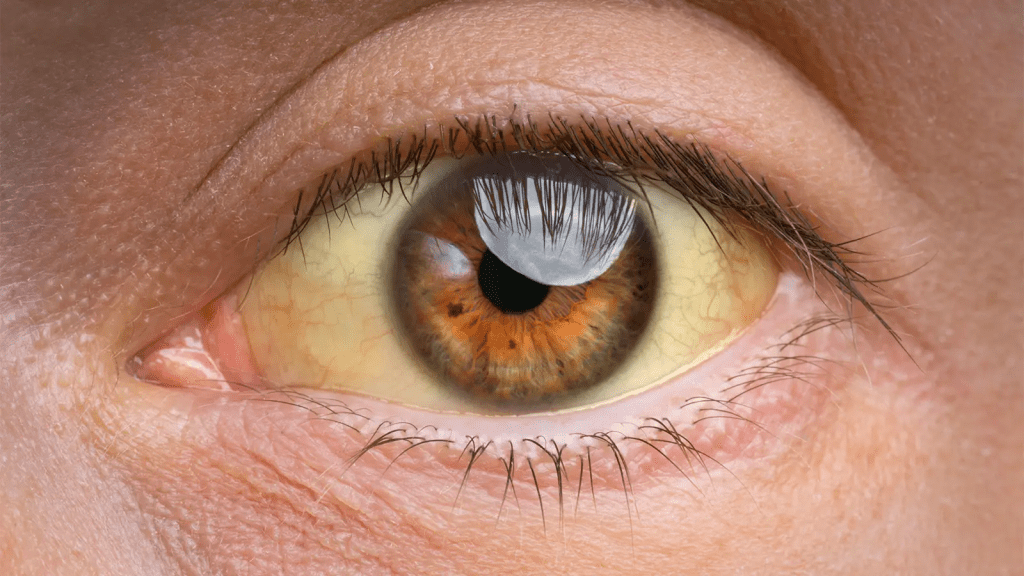
Muitas vezes os sintomas também se confundem com o que desencadeou o aumento da bilirrubina indireta e portanto a icterícia. Doenças das células vermelhas do sangue, como anemias hemolíticas, anemia falciforme, esferocitose e outras aumentam a taxa de destruição das hemácias e aumentam a quantidade de bilirrubina indireta para o fígado conjugar.
Mas os principais fatores desencadeantes da icterícia são aqueles que levam a piora temporária no funcionamento da enzima. São eles:
- Jejum prolongado
- Atividade física intensa
- Desidratação
- Stress
- Menstruação
- Abuso de álcool
- Falta de sono
- Infecções
- Cirurgias
- Período neonatal (bebês com Gilbert tendem a ter icterícia neonatal mais prolongada)
- Medicamentos (como a rifampicina)
DIAGNÓSTICO
O diagnóstico da síndrome de Gilbert começa com a demonstração do aumento da bilirrubina indireta no sangue. Ele costuma ser discreto, geralmente abaixo de 2,0 mg/dL, quase sempre abaixo de 4,0 mg/dL (na prática, pode subir um pouco mais, e a bilirrubina direta também pode estar um pouco acima do normal, por vários mecanismos, embora sempre menor que a indireta).
Diagnóstico de exclusão
Na prática, o próximo passo é excluir doenças que aumentem a produção de bilirrubina indireta (especialmente doenças hemolíticas) e descartar doenças do fígado. Para isso bastam ultrassonografia e exames laboratoriais simples. Se todos forem normais e a pessoa não tiver mais nenhum sintoma ou nada que sugira a presença de uma doença, isso é suficiente. Para uma condição extremamente comum e que não traz consequências futuras, é importante não arriscar problemas mais sérios com investigação invasiva.
Testes diagnósticos
Alguns testes usam a relação da enzima e da bilirrubina com outros fatores para fazer o diagnóstico. O jejum, ou reduzir a dieta na véspera, aumenta a bilirrubina. Medicamentos como a rifampicina vão aumentar a bilirrubina, outros como o fenobarbital vão diminuir. Isso levou a vários testes padronizados, sendo que sua utilidade é maior para a investigação científica do que na prática.

Teste genético
A pesquisa de mutações do gene UGT1A1 está disponível hoje em centros de pesquisa e grandes laboratórios, mas é caro. Geralmente só precisamos desse exame quando há alguma dúvida diagnóstica ou em pesquisas clínicas, onde é necessário a certeza ou em pesquisas farmacêuticas onde a presença da mutação pode alterar os resultados.
TRATAMENTO E PROGNÓSTICO
De modo geral, não há necessidade de tratamento para a síndrome de Gilbert. A icterícia geralmente é leve e temporária, mas recorrente. O que leva alguns portadores a procurar atendimento médico por questões estéticas. Nesse caso, é possível o uso de fenobarbital, mas isso geralmente não é recomendado pelos efeitos colaterais.

Como a UDP glucuronil transferase não faz a conjugação só da bilirrubina, mas também de diversos medicamentos, o metabolismo de algumas drogas pode ser um pouco diferente nos portadores da síndrome. Estatinas, usadas para reduzir o colesterol, podem desencadear aumento nas bilirrubinas e a medicação pode causar mais efeitos colaterais do que nos não portadores, mas isso não contra indica o tratamento. Do mesmo modo, há maior risco de intoxicação e lesão hepática pelo paracetamol e rifampicina, mas isso só é observado em doses acima das recomendadas.
A longo prazo, não há consequências sérias para o portador da síndrome, o aumento crônico de bilirrubina é considerado praticamente inócuo, mas observa-se um aumento no risco de pedras na vesícula. Por outro lado, por mecanismos ainda não bem conhecidos, há uma redução na mortalidade geral por câncer, especialmente de endométrio e linfoma de Hodgkin, e na aterosclerose, incluindo aí redução no risco de cardiopatia isquêmica. Acho que compensa ficar com os olhos um pouco amarelados de vez em quando por algum tempo, não é ?
CONCLUSÃO
A síndrome de Gilbert é extremamente comum, só não vemos mais diagnósticos porque a icterícia costuma ser muito leve, aparece só de vez em quando e dura poucos anos. O diagnóstico geralmente é feito excluindo outras doenças que levem ao aumento de bilirrubina indireta, mas há testes mais específicos disponíveis se houver alguma dúvida. O portador não necessita de nenhum tratamento e deve ser orientado em relação à benignidade da sua condição.
BIBLIOGRAFIA
- Nazer, Hisham. Unconjugated Hyperbilirubinemia Clinical Presentation. Medscape. Acessado em 21/05/23 (link)
- Roy-Chowdhury, J, Roy-Chowdhury, N e Wang, X. Gilbert syndrome and unconjugated hyperbilirubinemia due to bilirubin overproduction. Uptodate. Acessado em 21/05/23 (link)
- Gilbert’s syndrome. NHS. Acessado em 21/05/23 (link)
- Thoguluva Chandrasekar V, Faust TW, John S. Gilbert Syndrome. [Updated 2023 Feb 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470200/
- VETURIANO, M. S. .; CARVALHO FILHO, E. A. de .; RAMOS, M. G. C. . Gilbert syndrome: Literature review directed at diagnostic methods. Research, Society and Development, [S. l.], v. 12, n. 4, p. e6412440829, 2023. DOI: 10.33448/rsd-v12i4.40829. Disponível em: https://rsdjournal.org/index.php/rsd/article/view/40829. Acesso em: 21 may. 2023. (link)
Artigo criado em 21/05/2023
Última atualização: 22/05/23


