

INTRODUÇÃO
A alfa-1 antitripsina (A1AT) é uma proteína essencial produzida quase exclusivamente pelo fígado e liberada na circulação para proteger diversos tecidos (especialmente os pulmões) contra a ação de enzimas inflamatórias, como a elastase dos neutrófilos. Em condições normais, ela funciona como um “freio” natural da inflamação: quando um processo inflamatório ocorre, a A1AT neutraliza enzimas que, embora úteis para combater infecções, podem danificar o tecido saudável se agirem de forma descontrolada.

A deficiência de alfa-1 antitripsina (DA1AT) é uma condição genética hereditária causada por mutações no gene SERPINA1, localizado no cromossomo 14. Esse gene codifica a estrutura da A1AT, portanto alterações em sua sequência podem levar à redução na produção da proteína ou à síntese de uma forma anormal que não consegue sair adequadamente do fígado. Entre as várias variantes conhecidas, a mutação Z (Pi*Z) é a mais comum e clinicamente relevante: ela faz com que a proteína se dobre de forma incorreta, acumulando-se dentro dos hepatócitos (as células do fígado) em forma de aglomerados ou polímeros.
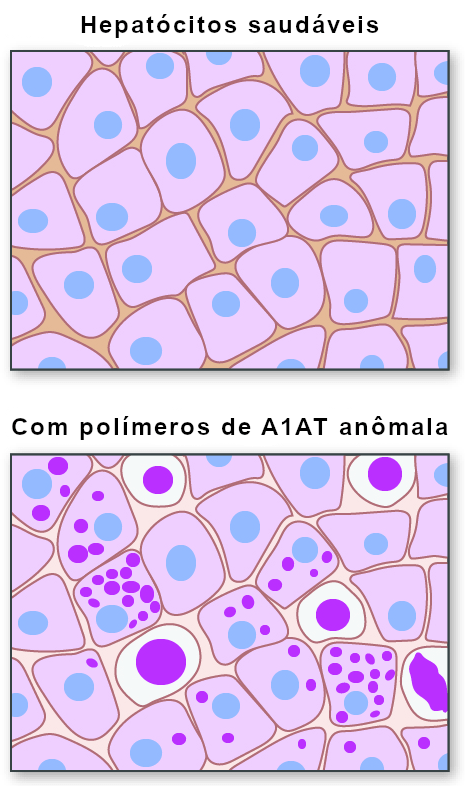
Com o tempo, esse acúmulo intracelular gera estresse celular, inflamação crônica e fibrose, danificando progressivamente o fígado. Além disso, como a proteína defeituosa não chega aos pulmões em quantidade suficiente, o indivíduo fica mais vulnerável a enfisema pulmonar e bronquite crônica. Assim, a A1ATD é uma doença sistêmica, com manifestações predominantes no fígado e nos pulmões — embora o impacto hepático seja particularmente relevante em crianças e em adultos expostos a fatores adicionais como álcool, obesidade e esteatose metabólica.
A principal manifestação da DA1AT é pulmonar, mas nesse texto vamos focar em como a doença pode danificar o fígado, que em parte dos casos é o principal órgão afetado.

O espectro clínico é amplo: algumas pessoas descobrem a deficiência apenas por acaso, em exames de rotina; outras evoluem com elevação persistente de enzimas hepáticas, fibrose, cirrose ou até hepatocarcinoma (HCC). Em adultos, a A1ATD é reconhecida hoje como uma das principais causas genéticas de doença hepática crônica, frequentemente subdiagnosticada devido à sua apresentação silenciosa e inespecífica.
EPIDEMIOLOGIA
A deficiência de alfa-1 antitripsina (DA1AT) é uma das doenças genéticas graves mais comuns entre pessoas de origem europeia, embora ainda seja pouco reconhecida e frequentemente subdiagnosticada. Estima-se que, em todo o mundo, cerca de 1 em cada 2.000 a 5.000 indivíduos tenha o genótipo Pi*ZZ, a forma homozigota e mais grave da deficiência. Isso corresponde a aproximadamente 230 a 250 mil pessoas afetadas globalmente — número comparável à prevalência de doenças raras conhecidas, mas com impacto muito maior devido ao grande contingente de portadores assintomáticos não diagnosticados.
As maiores prevalências ocorrem no norte da Europa (particularmente na Escandinávia, Reino Unido, Irlanda e Benelux), com frequência média do alelo Z entre 1 e 2%. À medida que se afasta dessas regiões, a frequência diminui, mas a variabilidade é grande — em parte porque a DA1AT é historicamente subdiagnosticada em países de alta miscigenação genética, como o Brasil. Estudos recentes indicam que as Américas concentram cerca de 35 a 40% dos casos mundiais, distribuídos de forma desigual conforme a ascendência europeia das populações locais.
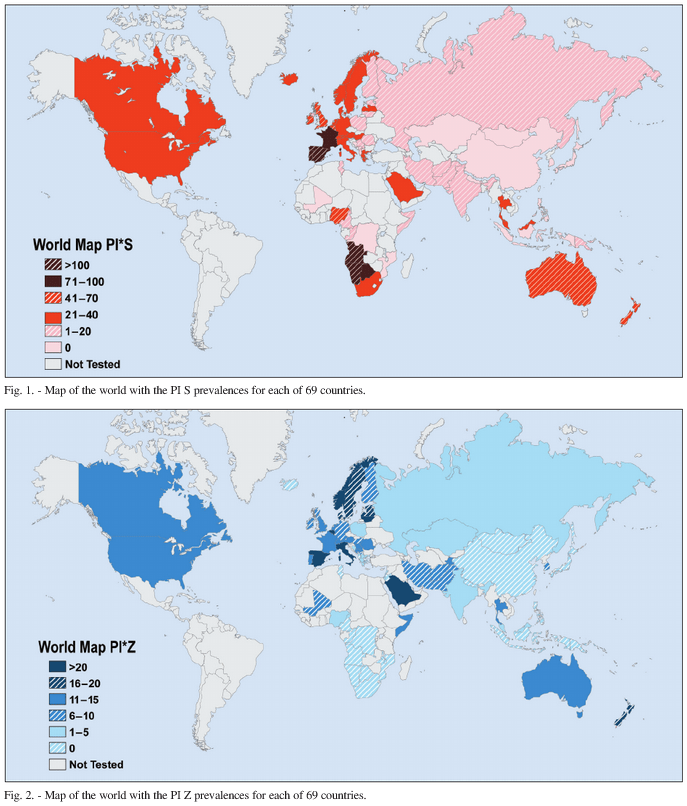
No Brasil, a verdadeira magnitude ainda é pouco conhecida, mas análises genéticas em bancos populacionais sugerem frequência do alelo Z entre 0,5 e 1%, o que se traduz em milhares de pessoas com formas graves (Pi*ZZ) e milhões de portadores heterozigotos (Pi*MZ). Apesar disso, menos de 10% dos casos são formalmente diagnosticados, segundo estimativas baseadas em registros laboratoriais e estudos da Alpha-1 Foundation e sociedades médicas nacionais.
Os portadores heterozigotos Pi*MZ são especialmente numerosos — possivelmente 1 a cada 25 pessoas em países de origem europeia e entre 2 a 5% da população brasileira, dependendo da região. A maioria não desenvolve doença clínica, mas esse grupo tem risco aumentado de lesão hepática quando coexistem fatores adicionais como esteatose, consumo de álcool, hepatites virais crônicas ou uso prolongado de medicamentos tóxicos. Em estudos recentes, o genótipo MZ foi duas a três vezes mais prevalente entre pacientes com cirrose alcoólica ou esteato-hepatite metabólica do que na população geral.
Além disso, há variantes raras (como null, I, F, Mmalton) que podem causar deficiência grave mesmo com níveis séricos aparentemente normais. Esses casos representam uma minoria (<1%), mas reforçam a importância de uma avaliação genética completa quando há suspeita clínica.
Um aspecto importante da epidemiologia moderna é que as manifestações hepáticas e pulmonares não se sobrepõem necessariamente: muitas pessoas com DA1AT grave têm fígado comprometido, mas pulmões normais, e vice-versa. Isso explica por que a hepatologia tem assumido papel central no rastreamento e manejo da DA1AT — especialmente no contexto da epidemia de doença hepática metabólica.
FISIOPATOGENIA
O fígado é uma verdadeira fábrica de proteínas, e entre elas está a alfa-1 antitripsina (A1AT) — uma molécula cuja principal missão é proteger tecidos como o pulmão da inflamação exagerada. Na deficiência de A1AT, especialmente nas formas ligadas ao alelo Z (Pi*Z), ocorre uma falha no processo de “dobramento” da proteína dentro do hepatócito.
1. O erro de fabricação
A mutação E342K (que caracteriza o alelo Z) faz com que a proteína A1AT seja sintetizada com estrutura tridimensional instável. Em vez de ser corretamente dobrada e exportada do retículo endoplasmático (RE) do hepatócito para o sangue, ela se aglomera dentro do RE, formando polímeros — pequenas “bolhas” de A1AT anormal, visíveis ao microscópio como glóbulos eosinofílicos (os “globos PAS positivos”).
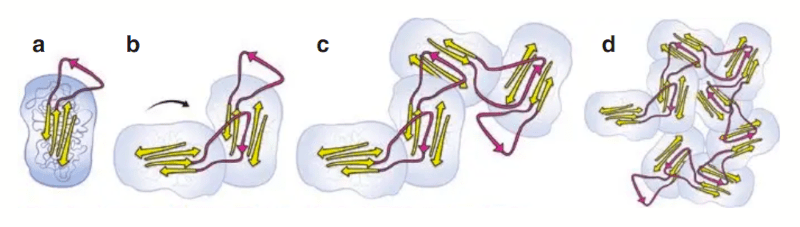
Esses agregados não conseguem ser eliminados adequadamente nem pelo sistema de degradação de proteínas (proteassoma) nem pela autofagia. O resultado é um engarrafamento celular — o fígado tenta produzir, mas não consegue despachar, gerando estresse do retículo endoplasmático, inflamação e, ao longo do tempo, morte celular (apoptose).
2. A cascata inflamatória e fibrogênese
Os hepatócitos sobrecarregados liberam sinais de alarme (citocinas, radicais livres e proteínas de estresse) que ativam as células estreladas do fígado, responsáveis pela deposição de colágeno. Com o tempo, isso leva à fibrose progressiva — primeiro leve e focal, depois mais difusa, culminando em cirrose. O processo é lento e muitas vezes silencioso por décadas: o fígado se regenera e inflama repetidamente até perder sua arquitetura normal.
Curiosamente, apenas uma parcela (≈10%) dos adultos com genótipo Pi*ZZ desenvolve cirrose clínico-radiológica, sugerindo que fatores adicionais (como álcool, esteatose metabólica, hepatite viral e polimorfismos genéticos moduladores) influenciam fortemente a progressão.
3. O duplo impacto: falta nos pulmões, excesso no fígado
Enquanto o fígado sofre pelo acúmulo tóxico, os pulmões sofrem pelo déficit funcional da proteína. Nos pulmões, a A1AT atua como “escudo” contra a elastase dos neutrófilos; sem ela, a elastina é degradada e o tecido pulmonar perde elasticidade, levando ao enfisema precoce. Mas no fígado, o problema é o oposto: acúmulo em excesso, e não carência.
4. A base genética e as variantes
O gene SERPINA1 apresenta centenas de variantes, mas as principais são:
- M (normal) — estrutura correta, exportação eficiente.
- S (E264V) — leve instabilidade, deficiência discreta.
- Z (E342K) — instabilidade grave, tendência à polimerização.
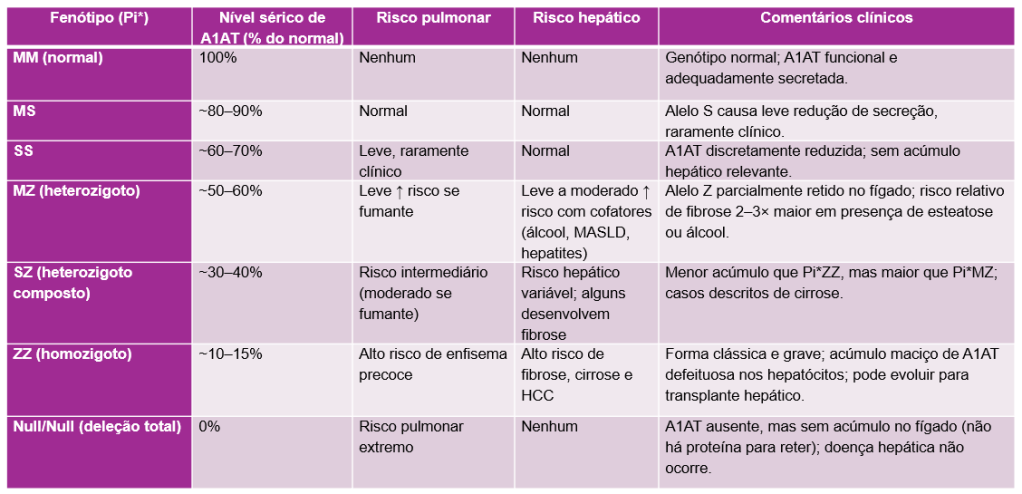
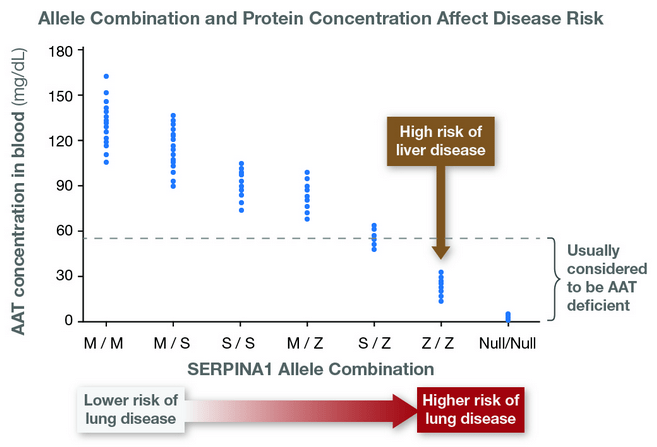
O alelo Z é codominante: isso significa que indivíduos Pi*MZ produzem tanto proteína normal quanto anormal. A quantidade total de A1AT circulante nesses casos é intermediária (cerca de 60%), mas ainda suficiente para proteger o pulmão — o problema é que mesmo pequenas quantidades da forma anormal já podem se acumular no fígado, especialmente quando há esteatose, álcool ou resistência à insulina, que aumentam o estresse oxidativo e prejudicam a “limpeza” celular.
5. O elo com o câncer de fígado
Com a progressão da fibrose, o risco de hepatocarcinoma (CHC) aumenta. Estudos mostram que o risco relativo de CHC em indivíduos Pi*ZZ é de 5 a 10 vezes maior do que em pessoas sem DA1AT, mesmo após ajuste para outros fatores. Acredita-se que isso ocorra por inflamação crônica, regeneração acelerada e acúmulo de proteínas mutadas que alteram a estabilidade genômica.
SINTOMAS (DO FÍGADO)
A doença costuma evoluir de forma silenciosa no fígado. Durante muitos anos — às vezes por décadas — o portador pode não apresentar sintomas perceptíveis, e a doença é descoberta por acaso, em exames de rotina.
Fase inicial (assintomática ou leve)
- Em grande parte dos casos, as primeiras alterações aparecem nos exames de sangue, com discreto aumento das enzimas hepáticas (AST, ALT e GGT).
- Muitas vezes, o paciente sente fadiga ou desconforto abdominal leve, mas não dá para dizer se os sintomas são causados pela doença ou por algum outro motivo que os leva a realizar exames e a detectar alterações no fígado.
Fase avançada (cirrose e hipertensão portal)
- À medida que a doença progride e compromete o funcionamento do fígado, surgem sinais mais claros de cirrose:
- Icterícia (coloração amarelada da pele e dos olhos);
- Ascite (acúmulo de líquido no abdome, fazendo o ventre aumentar de volume);
- Inchaço nas pernas (edema);
- Cansaço intenso e perda de massa muscular;
- Varizes de esôfago ou estômago, que podem causar sangramentos;
- Em casos avançados, confusão mental (encefalopatia hepática) e coceira intensa (colestase).
Esses sinais refletem insuficiência da função hepática e hipertensão portal, condições que exigem acompanhamento especializado e, em alguns casos, avaliação para transplante de fígado.
DIAGNÓSTICO
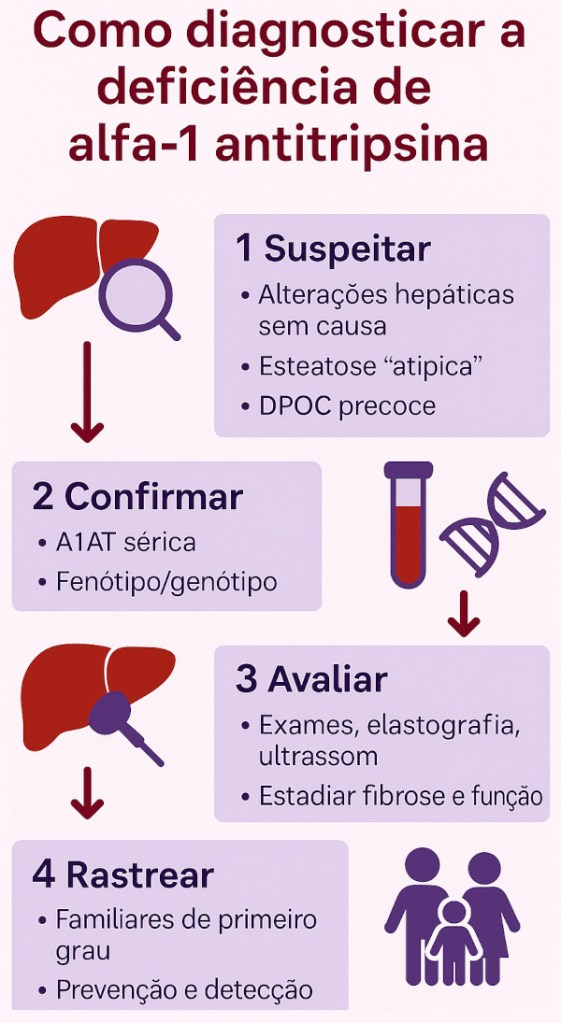
O diagnóstico da deficiência de alfa-1 antitripsina (DA1AT) exige atenção clínica e confirmação laboratorial — e pode ser surpreendentemente simples quando lembrado no raciocínio diagnóstico. A maior dificuldade não é o exame, mas suspeitar da doença.
1. Suspeitar do diagnóstico
A A1ATD deve ser lembrada sempre que o paciente apresentar:
- Alteração persistente das enzimas hepáticas (AST, ALT ou GGT) sem causa evidente (hepatite viral, álcool, autoimune etc.);
- Esteatose ou esteato-hepatite em pessoas sem obesidade ou síndrome metabólica, especialmente jovens;
- Cirrose criptogênica (sem causa aparente após investigação básica);
- História familiar de cirrose, hepatocarcinoma ou doença pulmonar precoce;
- Doença pulmonar obstrutiva crônica (DPOC), asma grave ou enfisema precoce (<45 anos), especialmente em não fumantes;
- Elevação desproporcional de GGT ou redução inexplicada de A1AT sérica em exames de rotina;
- Em crianças e adolescentes, episódios de icterícia prolongada ou elevação persistente de transaminases.
🔎 Em adultos, o “padrão típico” que deve acender o alerta é: homem ou mulher magro(a), com esteatose inexplicável e enzimas flutuantes, com ou sem histórico de asma/enfisema na família.
2. Confirmar com exames laboratoriais
a) Dosagem sérica da A1AT
- É o teste inicial de triagem.
- Valores <80 mg/dL (ou <11 µM) sugerem deficiência; <50 mg/dL são altamente sugestivos de formas graves (ex.: Pi*ZZ).
- Atenção: a A1AT é uma proteína de fase aguda — pode subir em infecções, inflamação ou gestação, levando a resultados falsamente normais. Por isso, é essencial confirmar com testes genéticos ou fenotípicos ou repetir após resolução da inflamação.
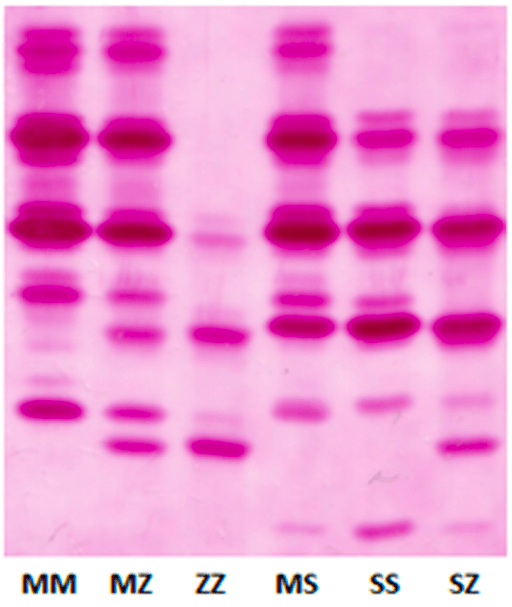
b) Fenotipagem (Pi)*
- Feita por focalização isoelétrica, identifica o padrão de migração das proteínas (ex.: Pi*MM, MZ, ZZ).
- Historicamente, antes mesmo da identificação do gene SERPINA1, pesquisadores observaram diferenças eletroforéticas entre as proteínas A1AT do sangue de diferentes pessoas. Essas formas variantes migravam com velocidades distintas em gel de amido, e foram denominadas de acordo com sua mobilidade eletroforética: Pi*M (M de “medium” ou “médio”): é a forma normal, com mobilidade intermediária. Pi*Z (Z de “slow”): forma anormal que se desloca mais lentamente no gel, por causa de uma alteração estrutural. Pi*S (S de “slow”, mas menos acentuado): variante também mais lenta, mas com menor impacto clínico que a Z. O termo “Pi” vem de Protease inhibitor (ou “inibidor de protease”) — que é justamente a função principal da alfa-1 antitripsina, e acabou sendo mantido em parte dos textos por questão de tradição. (figura de Carroll, 2014).
- É o método clássico e amplamente disponível em laboratórios especializados.
- Mostra o fenótipo funcional, mas não detecta variantes raras.
c) Genotipagem (SERPINA1)
- Detecta mutações específicas (Z = E342K, S = E264V, e variantes nulas ou raras).
- Pode ser feita por PCR, sequenciamento ou painéis genéticos.
- Confirma o diagnóstico e diferencia heterozigotos compostos (ex.: SZ, MZ, null/Z).
- Hoje, é o padrão ouro em centros de referência, podendo ser solicitada mesmo após triagem sérica normal quando há suspeita clínica forte.
3. Avaliar a repercussão hepática
Após confirmar a A1ATD, o passo seguinte é avaliar a extensão do dano hepático, especialmente em adultos.
a) Exames laboratoriais básicos
- TGO (AST), TGP (ALT), GGT, FA, bilirrubinas, albumina e RNI — para avaliar inflamação e função hepática.
- Plaquetas e relação AST/ALT ajudam a estimar risco de fibrose.
b) Métodos não invasivos de estadiamento
- FIB-4 (índice simples baseado em idade, AST, ALT e plaquetas).
- Valores <1,3 sugerem ausência de fibrose significativa.
- Valores >2,67 indicam fibrose avançada provável.
- Elastografia hepática (ou FibroScan®) — mede a rigidez hepática, estimando o grau de fibrose do fígado, a probabilidade de cirrose e de hipertensão portal.
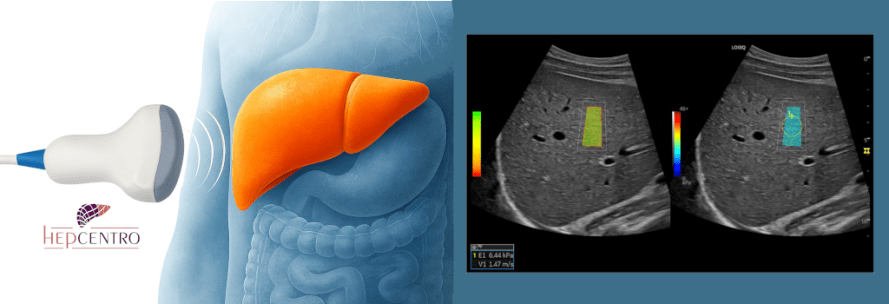
- Ultrassonografia hepática — avalia morfologia e, se houver cirrose, é usada para rastreamento de carcinoma hepatocelular (CHC) a cada 6 meses.

c) Biópsia hepática (casos selecionados)
- Indicada quando há discordância entre exames ou suspeita de outra doença associada.
- Mostra os “glóbulos PAS positivos”, típicos da retenção de A1AT no retículo endoplasmático.
- É hoje menos necessária graças aos métodos não invasivos, mas ainda útil em casos complexos.
TRATAMENTO HOJE
1. Não há terapia curativa direta para o fígado
Até o momento, não existe tratamento com eficácia comprovada capaz de remover ou corrigir a proteína A1AT defeituosa que se acumula dentro dos hepatócitos. Por isso, o manejo clínico concentra-se em prevenir novas agressões, controlar fatores de risco metabólicos e tratar complicações já instaladas.
2. Medidas gerais e prevenção da progressão
O cuidado de base é proteger o fígado. Medidas simples, porém fundamentais, têm grande impacto:
- Evitar álcool completamente, mesmo em pequenas quantidades.
- Controlar o peso corporal e tratar síndrome metabólica (diabetes, dislipidemia, hipertensão, apneia do sono).
- Adotar dieta equilibrada, semelhante à mediterrânea, rica em frutas, vegetais, fibras e gorduras monoinsaturadas.
- Atividade física regular, que melhora resistência à insulina e reduz inflamação hepática.
- Vacinar-se contra hepatites A e B e fazer rastreamento de hepatite C.
- Evitar medicamentos hepatotóxicos (como excesso de paracetamol, anabolizantes e alguns fitoterápicos).
3. Controle das comorbidades
A associação entre DA1AT e esteatose metabólica (MASLD) é hoje um dos principais fatores de progressão.
Por isso, o acompanhamento deve incluir:
- Avaliação endócrina e controle rigoroso de glicemia e lipídios;
- Monitoramento de apneia do sono, frequentemente negligenciada e pró-inflamatória;
- Acompanhamento nutricional para reduzir gordura visceral;
- Suporte psicológico para adesão à abstinência alcoólica e mudança de estilo de vida.
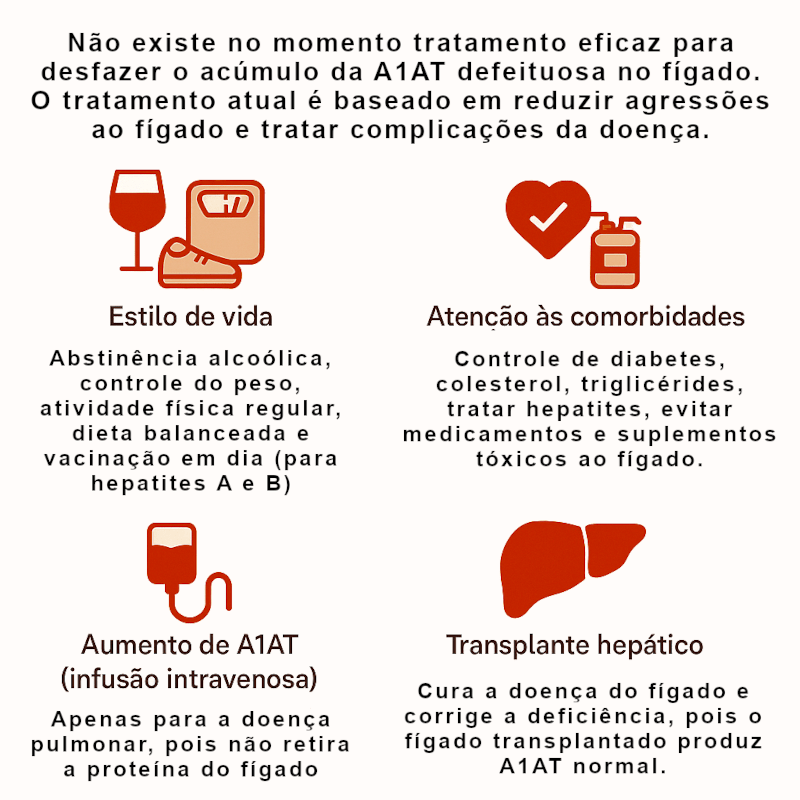
4. Terapia de reposição de A1AT (infusão intravenosa)
- Indicada somente para doença pulmonar associada (enfisema).
- O objetivo é elevar os níveis séricos de A1AT acima do limiar protetor pulmonar (≈ 11 µM).
- Não tem efeito hepático, pois não reduz o acúmulo da proteína defeituosa dentro dos hepatócitos.
- Estudos experimentais mostram ausência de benefício hepático e risco de sobrecarga adicional se usada indiscriminadamente.
5. Tratamento das complicações
Quando há cirrose, o manejo segue protocolos gerais de doença hepática avançada:
- Controle de ascite, varizes de esôfago, encefalopatia hepática;
- Rastreamento semelhante a outras causas de cirrose: ultrassom a cada 6 meses para hepatocarcinoma (CHC);
- Profilaxia e tratamento de infecções bacterianas em casos descompensados;
- Suplementação nutricional e vigilância de sarcopenia.
6. Transplante hepático
O transplante de fígado é atualmente o único tratamento curativo para a A1ATD hepática.
Ele substitui o fígado doente por um órgão que produz A1AT normal — curando tanto a doença hepática quanto a deficiência sistêmica.
- Indicado em cirrose descompensada, hepatocarcinoma dentro dos critérios de Milão, ou falência hepática aguda (que é rara nessa doença).
- Resultados são excelentes: sobrevida de >85% em 5 anos, e melhora pulmonar pela normalização da A1AT sérica.
- Não há recorrência da doença no fígado transplantado, pois o novo órgão tem o gene SERPINA1 normal.
PERSPECTIVAS
O tratamento da deficiência de alfa-1 antitripsina (DA1AT) está entrando em uma nova era, com terapias direcionadas à causa molecular da doença. Enquanto o manejo atual foca em proteger o fígado e tratar complicações, as abordagens experimentais buscam interromper o processo de acúmulo da proteína Z-AAT defeituosa — e, em alguns casos, até reverter a fibrose hepática.
1. Terapias de RNA de interferência (siRNA)
O maior avanço até o momento vem dos silenciadores gênicos de RNA (siRNA), que “desligam” parcialmente o gene SERPINA1 no fígado. O principal representante é o fazirsiran, desenvolvido pela Arrowhead Pharmaceuticals em parceria com a Takeda.
Como funciona:
- O siRNA é entregue diretamente ao fígado através de um conjugado de GalNAc, que reconhece os hepatócitos.
- Ele interrompe a produção da proteína Z-AAT mutante, diminuindo o acúmulo tóxico intracelular.
- Com menos proteína defeituosa, o hepatócito sofre menos estresse e inflamação — permitindo que mecanismos de reparo e degradação atuem de forma mais eficaz.
Evidências clínicas (até 2025):
- Em ensaios de fase 2 (NEJM 2022; Gastroenterology 2024), o fazirsiran reduziu em até 80–90% a concentração hepática de Z-AAT.
- Houve melhora histológica significativa, com redução dos glóbulos PAS+, diminuição da inflamação e sinais de regressão de fibrose em parte dos pacientes.
- O medicamento foi bem tolerado, com eventos adversos leves e transitórios.
- Ensaios de fase 3 (estudo AFFIRM-AT, em andamento) estão avaliando se essa melhora histológica se traduz em benefício clínico duradouro, incluindo prevenção de cirrose e HCC.
🔬 Importante: O fazirsiran não repõe a A1AT circulante — ele apenas impede a produção da forma tóxica. Por isso, pode ser combinado futuramente com terapias de reposição pulmonar.

2. Terapias de RNA de edição (RNA-editing)
Outra estratégia emergente é a edição seletiva de RNA, que visa corrigir o erro da mutação Z (Glu342Lys) diretamente no RNA mensageiro, sem alterar o DNA.
- Utiliza enzimas do tipo ADAR (adenosina-desaminase de RNA), que convertem o códon mutado em uma sequência normal.
- A vantagem é que não há modificação permanente no genoma, reduzindo riscos.
- Estudos pré-clínicos em modelos murinos já mostraram restauração parcial da secreção de A1AT normal e redução do acúmulo intracelular.
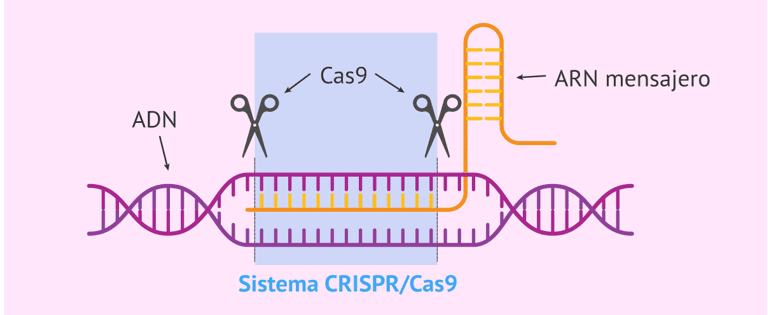
3. Edição gênica (CRISPR/Cas9 e base editing)
Pesquisas ainda iniciais utilizam sistemas de edição de DNA como o CRISPR/Cas9 ou o base editing, para corrigir permanentemente a mutação no gene SERPINA1.
- Modelos em camundongos mostraram produção estável de A1AT normal e eliminação completa do acúmulo hepático após a correção.
- Desafios atuais: segurança, eficiência de entrega (vetores virais ou nanopartículas) e controle da resposta imune.
- Ensaios clínicos em humanos ainda não iniciaram (previstos para 2026–2027).
4. Terapias de “chaperonas” farmacológicas
Uma chaperona é uma proteína ou molécula que auxilia outras proteínas a se dobram corretamente, se montarem e se transportarem dentro da célula, garantindo sua função adequada e a saúde celular. No contexto da DA1AT, essas moléculas pequenas visam melhorar o dobramento da A1AT mutante dentro do retículo endoplasmático, reduzindo a tendência de polimerização.
- Resultados experimentais mostram redução do estresse celular e menor acúmulo de inclusões em hepatócitos humanos cultivados.
- Ainda em fase pré-clínica, mas pode se tornar uma abordagem complementar às terapias gênicas.
BIBLIOGRAFIA
- Alpha-1 Antitrypsin Deficiency. Learn.Genetics (link)
- Bergasa, N.V. (2022). Alpha-1-Antitrypsin Deficiency. In: Bergasa, N.V. (eds) Clinical Cases in Hepatology. Springer, London. https://doi.org/10.1007/978-1-4471-4715-2_10
- Lopes AP, Mineiro MA, Costa F, Gomes J, Santos C, Antunes C, Maia D, Melo R, Canotilho M, Magalhães E, Vicente I, Valente C, Gonçalves BG, Conde B, Guimarães C, Sousa C, Amado J, Brandão ME, Sucena M, Oliveira MJ, Seixas S, Teixeira V, Telo L. Portuguese consensus document for the management of alpha-1-antitrypsin deficiency. Pulmonology. 2018 Dec;24 Suppl 1:1-21. doi: 10.1016/j.pulmoe.2018.09.004. PMID: 30473034.
- Gonzalez, JJ. Why does alpha-1 antitrypsin deficiency cause liver disease? Liver Fellow Network (link)
- Carroll, Tomás P., M. Emmet O’Brien, Laura T. Fee, Kevin Molloy, Blair Murray, Seshma Ramsawak, O. Mcelvaney, Catherine A O’Connor and Noel G. McElvaney. “Alpha-1 Antitrypsin Deficiency — A Missed Opportunity in COPD?” (2014).
- Stoller JK, Brantly M. The challenge of detecting alpha-1 antitrypsin deficiency. COPD. 2013 Mar;10 Suppl 1:26-34. doi: 10.3109/15412555.2013.763782. PMID: 23527684.
- de Serres FJ, Blanco I, Fernández-Bustillo E. PI S and PI Z alpha-1 antitrypsin deficiency worldwide. A review of existing genetic epidemiological data. Monaldi Arch Chest Dis. 2007 Dec;67(4):184-208. doi: 10.4081/monaldi.2007.476. PMID: 18309698.
- Villar, L. P. Empieza el primer proyecto de modificación genética de embriones humanos. Reproducción Asistida ORG. 2020. Disponível em: <https://www.reproduccionasistida.org/autorizacion-de-modificacion-de-embriones/> Acesso em: Acesso em 11 jun 2021.
- Alpha-1 Foundation / J Bras Pneumol 2024: materiais de apoio ao diagnóstico, educação e epidemiologia na América Latina/Brasil.
- AASLD – Liver Fellow Network (Why Series): visão clara da fisiopatologia e dados de risco de cirrose/HCC na vida adulta. aasld.org
- Fromme M, et al. 2025 (Hepatology Communications & review em acesso livre): revisão abrangente e atualizada da A1ATD hepática, com estratificação não invasiva e fatores de progressão. PMC+1
- Blanco I, et al. 2023: estimativa global de prevalência do genótipo Pi*ZZ. PubMed
- Holinka M, et al. 2025: estudo que relaciona MZ com maior frequência em cirrose por álcool/MASLD — importante para aconselhamento de heterozigotos. PMC
- EASL 2024 – Diretrizes de colestases genéticas: recomenda dosar A1AT como “triagem” para A1ATD e confirmar com fenótipo/genótipo. ScienceDirect
- NEJM 2022 – Fazirsiran (Strnad P, et al.) e Gastroenterology 2024 – Fazirsiran (Clark V, et al.): evidências clínicas de redução de Z-AAT, melhora histológica e sinal de regressão de fibrose; base das perspectivas terapêuticas. The New England Journal of Medicine+1
- Patel D, 2021 (CMAJ/PMC): aumento de A1AT não é indicado para doença hepática por A1ATD. PMC
- AASLD – Diretrizes HCC: rastreamento semestral com US em cirróticos (independente da causa). aasld.org
Artigo criado em: 24/10/25
Última atualização: 25/10/25


